Article Text

Abstract
Background Aicardi-Goutières syndrome (AGS) is a genetic inflammatory disorder that presents with early infantile encephalopathy. We report the clinical and molecular details of multiple members of a family with AGS secondary to a novel RNASEH2C mutation, highlighting the evolution of phenotypic abnormalities in AGS.
Methods Between February 2018 and June 2019, a pedigree tree was constructed for 141 members of a family. The clinical and radiological details of 14 symptomatic children were chronicled and compared with the asymptomatic family members. Genetic analysis was performed on 23 individuals (six symptomatic). This involved whole exome sequencing for one patient and confirmation of the identified indel variant in other family members.
Results The symptomatic children were diagnosed as AGS secondary to a novel indel variation in exon 2 of the RNASEH2C gene (chr11:65487843_65487846delinsGCCA). Clinically, between the ages of 2 and 6 months, the symptomatic children developed irritability (14/14), unexplained fever (9/14), chill blains (12/14), sleep irregularities (14/14) and developmental delay (14/14), with deterioration to vegetative state at a median (IQR) age of 10.5 months (9.25–11). In addition, chill blains were observed in 5/17 (29.4%) carrier individuals. Neuroimaging demonstrated a gradual progression of calcification involving basal ganglia, periventricular white matter and dentate nucleus. Three patients also demonstrated presence of subependymal germinolytic cysts.
Conclusion This report highlights a novel founder RNASEH2C mutation and the phenotypic evolution of AGS. In addition, we report chill blains in one-third of RNASEH2C mutation carriers. Neuroradiologically, the report illustrates novel MRI findings and demonstrates a progression pattern of disease. These findings will aid in earlier suspicion and diagnosis of AGS.
- cerebral palsy
- genetics
- paediatric neurology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
We studied members of a large family who had multiple individuals diagnosed as ‘cerebral palsy’. Cerebral palsy (CP) is a static encephalopathy occurring secondary to an insult to a developing brain. The affection of multiple family members with CP suggested a CP mimic with possible genetic basis. The subsequent investigations of affected family members led to a diagnosis of Aicardi-Goutières syndrome (AGS; OMIM225750), secondary to a novel RNASEH2C mutation.
AGS is a genetic inflammatory disorder caused due to mutations in one of the seven genes (AGS1-7).1 2 These genes regulate the nucleic acid metabolism resulting in intracellular overload of nucleic acids that leads to activation of TLR-Interferon-⍺ pathways.3 4 Clinically and radiologically AGS has features overlapping with CP and with congenital infections, which prompted the earlier terminology of ‘pseudo TORCH’.5 AGS is suggested by the clinical–radiological features and confirmed by genetic testing.
We studied the clinical details of 14 affected family members (six genetically confirmed) and compared them with the unaffected individuals of the family to define the clinical and radiological characteristics of AGS.
Methods
Patients
After obtaining informed consent, each family member underwent review of medical history and physical examination. AGS phenotype was defined as: (a) no antenatal adverse events; (b) no perinatal or neonatal complications; (c) developmental delay with onset before 6 months of age; and (d) increased tone. The family members after review of medical record underwent evaluation for neurological symptoms and examination was performed. The investigations including neuroimaging (MRI brain and/or CT brain) were chronicled. If neuroimaging was not available, then MRI brain was performed for the individuals fulfilling the AGS phenotype criteria. CSF analysis could be performed in only one patient, while TORCH titres could be done in two patients. The study was carried out at a tertiary care centre in India between February 2018 and June 2019.
Genetic testing and classification
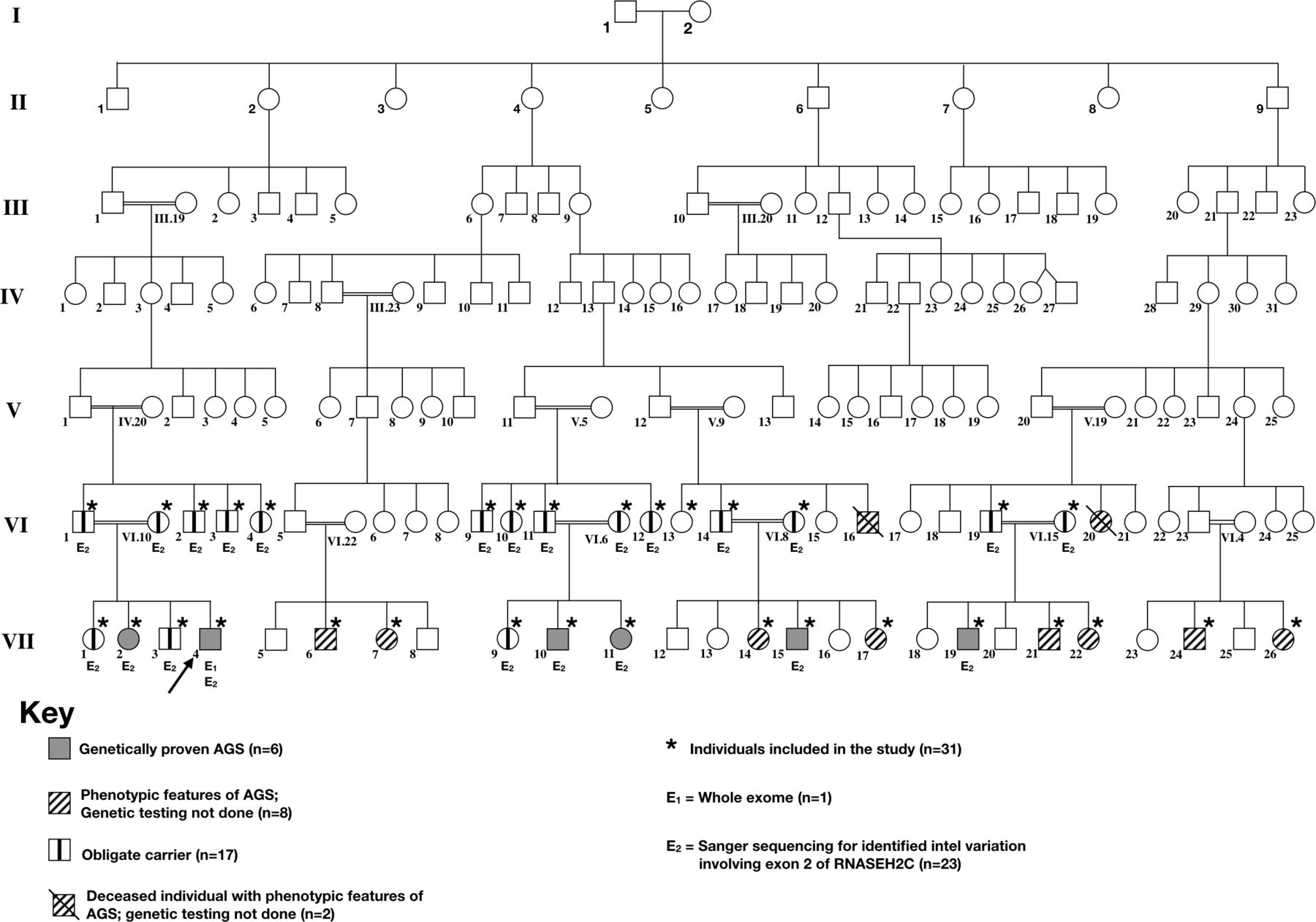
After obtaining informed consent detailed phenotypic classification was carried out and genetic testing was offered. A pedigree chart (figure 1) for the family was constructed in accordance with guidelines of Pedigree Standardisation Task Force.6 The phenotypic abnormalities could be confirmed for 19 individuals, of whom 14 were alive. The primary patient who presented to our facility was case 1 (VII.4). After the clinical, laboratory and radiological evaluations, he underwent whole exome sequencing. The mutation identified on WES was confirmed by Sanger sequencing. Sanger sequencing was thereafter performed for other family members. Based on the same, individuals were categorised as: (a) affected individuals (n=6): who had AGS phenotype and had homozygous mutations; (b) possibly affected (n=8): included individuals who had clinical features suggestive of AGS but genetic testing could not be carried out; (c) obligate carriers (n=17): who did not have AGS phenotype but had heterozygous AGS mutation; (d) Unaffected individuals: who neither had AGS phenotype nor AGS mutation; and (e) unknown (n=9): who were clinically asymptomatic but genetic testing could not be performed.

Seven-generation pedigree of the affected family. Generation number is listed along the left margin, and individuals are numbered underneath each symbol. The proband (indicated by solid white arrows) corresponds to Patient VII.4. Solid-shaded symbols correspond to affected members who were confirmed for genetically confirmed Aicardi-Goutières Syndrome (AGS). Spouses and unknown or unaffected family branches are not shown.
Phenotypic details of affected individuals
The phenotypic details of genetically proven cases are as given below.
Case 1 (VII.4)
This 6-month-old male infant, with no antenatal or perinatal adverse events presented with delayed attainment of age appropriate milestones and tightness of all four limbs noticed since 3 months of age. He was born at term with a birth weight of 2.9 kg. At 6 months, his occipito-frontal circumference (OFC) was 35.5 cm. Neurological examination showed spasticity and brisk reflexes. His abdominal and ophthalmological assessment was normal. There was history of chill blains, which used to occur on exposure to cold environmental surroundings but used to be more severe than other family members who were not affected by disease. His CSF examination revealed mild pleocytosis (0.012 X 109 WBCs/L), with elevated proteins (82 mg/dL). His TORCH titres were negative and his EEG was normal.
Case 2 (VII.2)
This 8-year-old sister of case 1 was being managed as CP. She was evaluated for non-attainment of any developmental milestone. She was born at term with birth weight of 3.1 kg with no antenatal or perinatal complications. On examination, she had microcephaly (OFC=39.5 cm), spasticity, hyper-reflexia and chill blains. Her TORCH titres and EEG did not reveal any abnormality.
Case 3 (VII.10)
This 6-year-old boy, had neurodevelopmental delay and had not attained any developmental milestone. He was born at term with a birth weight of 3.4 kg and at 6 years, his evaluation revealed: OFC=36.5 cm, bradykinesia and hypertonia with hyper-reflexia, with history of chill blains.
Case 4 (VII.11)
This 2-year-old girl, born at term with a birth weight of 3.1 kg, with no antenatal or perinatal adverse event, had developmental delay with microcephaly (OFC=35.5 cm), hypertonia and hyper-reflexia, without any chill blains.
Case 5 (VII.15)
This 8-year-old boy was born at term to an uneventful pregnancy. The perinatal period was uneventful and parents reported normal psychomotor development till 3–4 months of age. From the age of 4 months, he started developing episodes of unexplained fever at a frequency of nearly one episode every month, which used to last for 5–7 days and required oral medications. These episodes persisted till 20–22 months of age. These episodes were associated with irritability. He also developed chill blains from an age of 4 months. Around 6 months, he developed psychomotor regression with loss of all motor skills and by the age of 8 months, his developmental age was less than 3 months. When assessed his head circumference 37 cm and his neurological evaluation was suggestive of spasticity and hyper-reflexia. There was history of chill blains in neurologically normal parents and siblings too.
Case 6 (VII.19)
This boy was born at 38th week of gestation with a birth weight of 2.9 kg. His antenatal and perinatal periods were uneventful. At fourth month, parents noticed increased irritability and sleep irregularities. At 5 months of age, he was evaluated for developmental delay and was noted to lack head control, palmar grasp and vocalisation. At the age of 7 months, he was diagnosed as CP. The child had irritability, spasticity and feeding difficulties.
Results
Pedigree analysis
A pedigree tree was constructed with available details of 141 family members spanning over seven generations of this family with North Indian ancestry (figure 1). This included six affected individuals, eight possibly affected and 12 obligate carriers. The disease was manifesting only in children of consanguineous marriage suggesting an autosomal recessive phenotype.
Clinical phenotype
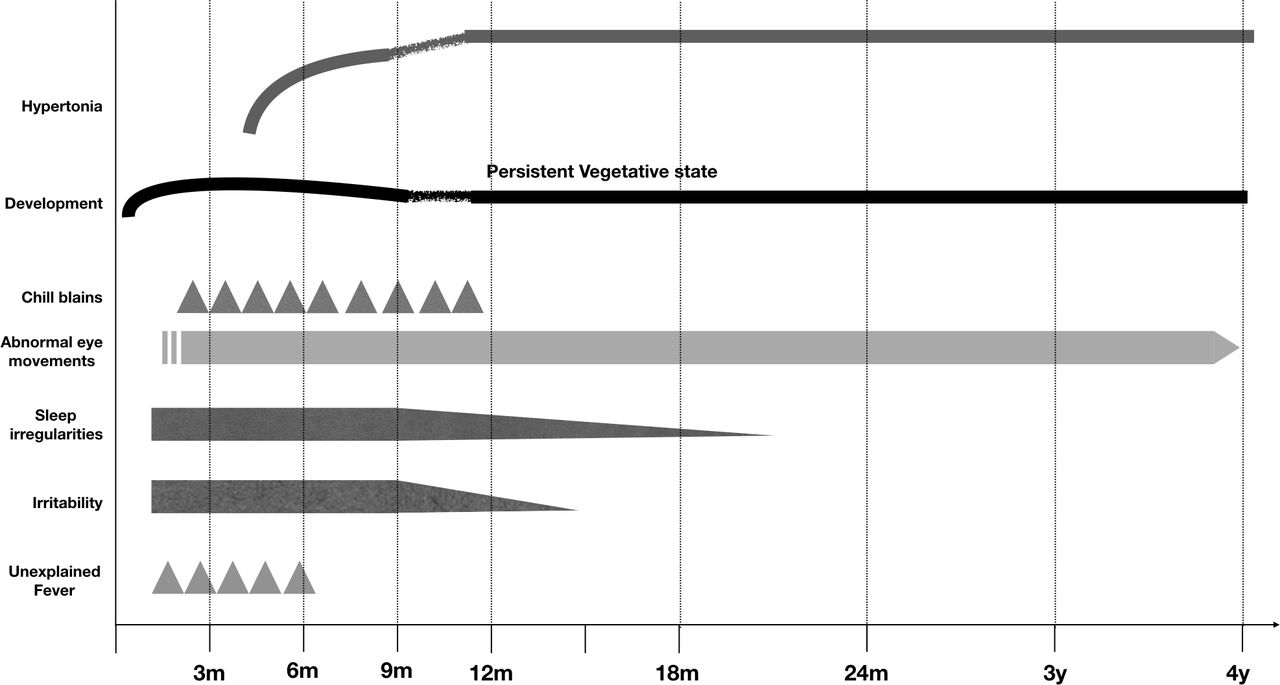
All children had an uneventful antenatal and perinatal period. All affected (n=6) and possibly affected individuals (n=8) became symptomatic before 3 months of chronological age (CA) with initial symptoms being irritability, sleep irregularities, unexplained fever, chill blains, abnormal eye movements and psychomotor delay. In addition, all symptomatic children had an OFC below 38 cm, suggesting head size had become static before 3 months of CA. Tonal abnormalities characterised by hypertonia (spasticity and/or rigidity) became prominent between 3 and 6 months of CA and it continued to worsen till 12 months of age, following which it stayed static for subsequent years. Between the ages of 6 and 24 months, the children had developed spasticity, bradykinesia and feeding difficulties. The clinical phenotype has been illustrated in table 1 and figure 2.
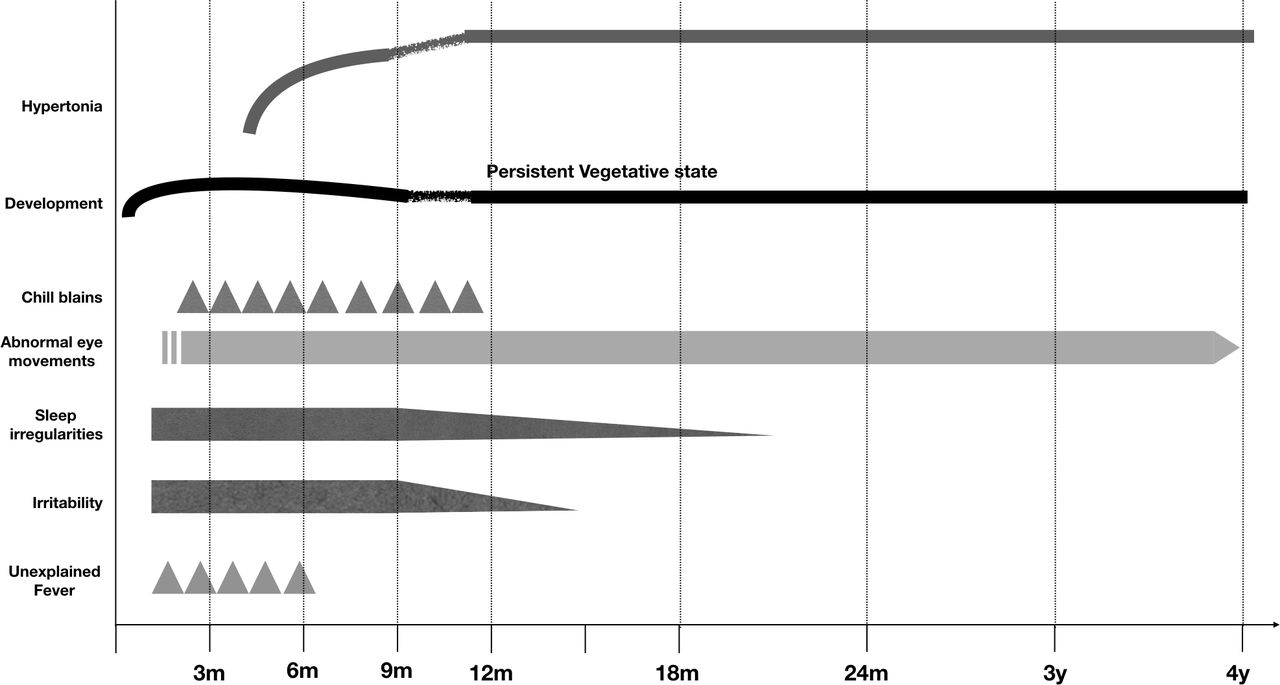
Graphical representation depicting the clinical course of children with Aicardi-Goutières Syndrome.
Clinical features of Aicardi-Goutières Syndrome (AGS)
Neuroradiology
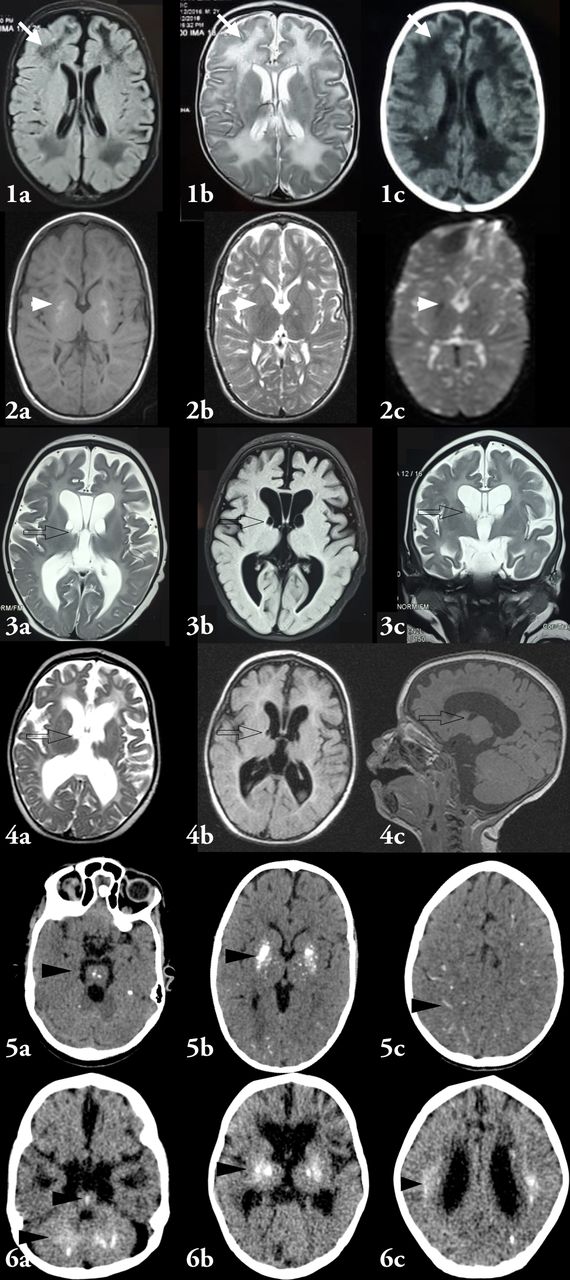
Neuroradiological characteristics have been highlighted in table 2 and figure 3. Neuroradiology was available for six affected AGS and seven possible AGS. All affected patients demonstrated basal ganglia calcification, cerebral atrophy and white matter changes. Intracranial parenchymal calcification pattern showed gradually increasing density of calcification in basal ganglia with advancing age. In addition, the calcification progressed with advancing age to involve striatum, thalamus, periventricular white matter, brainstem and dentate nucleus. Cerebral atrophy was demonstrated in all patients. However, in one patient (Case 2, VII.2), who underwent MRI at 8 years of age, MRI revealed only mild cerebral atrophy. Three patients also demonstrated subependymal germinolytic cysts.
{kind=link}
{kind=link}
{kind=link}
Neuroimaging characteristics of patients. Image 1 shows MRI findings of case VII.4, done at an age of 7 months. MRI shows diffuse white matter abnormality (white arrowheads) characterised by hyperintense signals on T2 and hypointense signals on T1. In addition, the ventricles are prominent and are suggestive of diffuse cerebral atrophy. Image 2 shows MRI findings of case VII.2 done at 8 years of age showing altered signal intensities in bilateral lentiform nuclei (hyperintense on T1 and isointense on T2/FLAIR) and showing signal blooming on GRE. No restriction of diffusion was observed. In addition, there was generalised prominence of ventricular system, sulcal and cisternal spaces suggestive of cerebral atrophy with predominant paucity of white matter especially in bilateral frontal lobes. However, the cerebral atrophy was mild and remarkably less as compared with other children. Image 3 illustrates MRI findings of case VII.10, done at 2 months of age. There is generalised prominence of ventricular system, and sulcal and cisternal spaces suggestive of diffuse cerebral atrophy. Subependymal germinolytic cysts (black outlined clear arrows) are seen in bilateral caudothalamic groove. There is diffuse white matter signal abnormality. Image 4 depicts the MRI findings of case VII.11 done at 6 months of age. The findings are similar to those listed for image 3. Image 5 illustrates NCCT findings of case VII.15 at 2 months of age at the onset of irritability. These are characterised by punctate calcifications involving brainstem (5A), globular and punctate calcifications involving putamen, globus pallidi and thalami. The cerebral atrophy however, is not prominent in this child. Image 6 depicts NCCT brain for case VII.19. This shows diffuse cerebral atrophy with paucity of white matter. Calcifications are noted in bilateral dentate nuclei and brainstem (6A), bilateral gangliocapsular regions and bilateral thalami (6B). In addition, punctate calcification is noted in bilateral periventricular white matter (6C). The calcifications have been indicated by black arrowheads. In none of the images, temporal lobe cysts are observed.
Radiological features of Aicardi-Goutières syndrome (AGS)
Genetics
Case 1 underwent whole exome sequencing. This demonstrated a homozygous indel variation in exon 2 of the RNASEH2C gene (chr11:65487843_65487846delinsGCCA) that resulted in the amino acid substitutions of Valine for Glutamic acid and Alanine for Valine at codons 72 and 73 respectively (p.Glu72_Val73delinsValAla; ENST00000308418). This RNASEH2C variant lies in the Ribonuclease H2 non-catalytic subunit (Ylr154p-like) domain of the RNASEH2C protein. This variant had not been reported in the 1000 genomes and has a minor allele frequency of 0.002% in the ExAC database. The in-silico prediction of the variant is benign by MutationTaster2. The variant was confirmed in case one by Sanger sequencing, which was followed by Sanger sequencing for aforementioned variant in five affected individuals (cases 2 to 6) and in 12 possible carriers. The affected individuals tested homozygous for the mutation, while the asymptomatic individuals were heterozygous for the same (obligate carriers). The chromatogram has been depicted in figure 3.
Discussion
AGS is a genetic inflammatory disease.2 We report the pedigree analysis of six individuals with genetically confirmed AGS, and eight children with phenotypic features of AGS, all belonging to the same family. The different age profiles of the patients allowed us to describe the evolution of clinical and neuroradiological characteristics of AGS. Additionally, we describe a novel mutation involving RNASEH2C gene affecting six individuals in homozygous state with 17 carriers and allowed us to compare the clinical features of affected individuals with the unaffected ones.
All 14 affected individuals suffered from progressive encephalopathy. These findings are consistent with earlier reports, where authors have described progressive encephalopathy with onset in first year of life.7 8 The encephalopathy progressed from an initial irritability and disturbance of sleep rhythms at an age of less than 3 months. This also coincided with episodes of sterile fever and stagnation of development. Between 3 and 6 months of age, irritability and sleep disturbances worsened before decreasing after 12–18 months of age. The developmental delay and encephalopathy worsened between 6 and 9 months of age and all the children gradually deteriorated to a behavioural vegetative state. Concomitantly, patients developed tonal abnormalities including spasticity, rigidity, bradykinesia and oromotor dyskinesia. These features were first noted around 5 months, and gradually worsened over infancy and stagnated thereafter. Before 6 months, bradykinesia and rigidity were dominant, while after 6 months of age hypertonia was uncharacterisable with features of both rigidity and spasticity at different points of examination. These findings corroborated with progressive changes noted on neuroradiology with basal ganglia calcification, white matter changes and cerebral atrophy, all setting in before 3 months. Gradually progressive generalised cerebral atrophy correlated with initial irritability, tonal abnormalities and progressing to vegetative state. These features, possibly highlight the progressive white matter and grey matter inflammation as part of disease.
We report three novel findings based on the analysis. First, literature has highlighted that nearly 40% of children with AGS may develop chill blains lesions involving fingers, toes and ears. In our study, chill blains were noted in 12 of 14 children (85.7%). These episodes occurred only on exposure to low environmental temperatures. In addition, five of 17 carriers also reported experiencing intermittent chill blains on exposure to cold temperature, though none of them had a severe or persistent skin lesion. Similar affection of AGS families has been reported based on unpublished survey by Rice et al.9 The explanation for the same is uncertain, though it may suggest associated systemic inflammation.
Second, three patients of the studied family had subependymal germinolytic cysts on MRI. These cysts have not been previously reported with AGS; though their association with congenital viral infections, metabolic disorders (especially Zellweger syndrome) and chromosomal abnormalities is reported in literature. Though, these cysts may be an isolated finding in otherwise healthy newborns, their occurrence in two patients suggests a possible ongoing vascular inflammation involving germinal matrix. This hypothesis however, will need further validation. In addition, none of our patients demonstrated temporal lobe cysts, which have been earlier associated with AGS.
Third, we demonstrate a novel mutation involving RNASEH2C gene. RNASEH2C gene interacts with RNASEH2A and RNASEH2B genes to form an enzymatic protein complex which cleaves ribonucleotides mis-incorporated into genomic DNA and resolves RNA:DNA duplexes.10 Mutations involving RNASEH2C gene are associated with impaired ribonucleotide excision repair and accumulation of embedded ribonucleotides.11 In addition, a recent report has highlighted that RNASEH2C knock-down up-regulates interleukin (IL)-6 and type 1 interferon beta suggesting the role of the gene in inflammation.12 The pathogenesis of AGS is immune mediated and inflammatory. RNASE2H deficient cells may lead to accumulation of immune reactive genetic material with consequently impaired suppression of IFN induction, similar to pattern seen with ADAR1 mutations.4 The earliest clinical features of irritability progressing to deteriorating encephalopathy support ongoing inflammatory brain damage. The initial selective damage involving germinal centres, and striatum in our patients suggests earlier involvement of metabolically active areas of brain.
The major limitation of this pedigree analysis is that we are unable to contribute to underlying pathogenesis of the disease because most of the specialised molecular studies were unavailable. Therefore, the hypothesis regarding role of systemic inflammation in chill blains (even in heterozygous individuals) and possible (inflammatory) vasculitis for germinolytic cysts needs to be confirmed with future observations involving molecular and serological studies. To conclude, our report demonstrates the evolution of clinical and radiological features of AGS, highlights the role of pedigree analysis in patients with suspected CP and also reports a novel AGS mutation. These findings might be useful in and earlier and precise clinical and radiological diagnosis of AGS.
Acknowledgments
The authors thank the families involved for their participation.
References
Footnotes
Contributors The clinical evaluation of patients was performed by DL, AS, GB and VS. Radiological evaluation was performed by UR and VS. DL, AS, GB and VS participated in pedigree construction and analysis. The initial draft of manuscript was prepared by VS and DL, which was edited by UR and VS. The final draft was approved by all authors.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data sharing not applicable as no datasets generated and/or analysed for this study. The study involves pedigree analysis. The details of pedigree are part of the study. No additional data was obtained/analysed.