Article Text

Abstract
Parkinsonism is seen frequently in patients with psychiatric conditions. Drug-induced parkinsonism (DIP) is the second most common cause of parkinsonism in the general population after Parkinson’s disease (PD) but a range of rarer aetiologies, some of them reversible, should also be considered in patients of all ages. DIP is more common in older patients, as are neurodegenerative diseases that may produce parkinsonism and it is relatively more likely that drug exposure could be unmasking an underlying process in this population. There is an extensive literature on differentiating DIP from PD but clinical features can be indistinguishable and many proposed investigations are not readily available. Aside from cessation of the responsible medication, there is no clear consensus on treatment strategies or duration of treatment. Practically, a delicate balance must be achieved between ameliorating parkinsonism and avoiding recurrent psychosis. Long-term prognosis in the setting of DIP remains unclear. We review the features that may differentiate DIP from other causes of parkinsonism in patients with psychiatric illness, provide an update on relevant investigations and discuss management strategies. The use of atypical antipsychotics for a broad range of indications highlights the ongoing relevance of DIP.
- parkinson's disease
- neuropsychiatry
- pharmacology
- psychiatry
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Parkinsonism, defined as bradykinesia in combination with either rest tremor, rigidity or both,1 classically arises in the context of Parkinson’s disease (PD). The major pathology is represented by Lewy bodies containing alpha-synuclein and associated dopamine cell loss in the substantia nigra leading to dysfunction of basal ganglia circuitry responsible for motor control. However, parkinsonism due to a wide range of possible causes also occurs in up to one-third of patients taking antipsychotic medications.2 This syndrome may significantly impair function and quality of life.
Pathophysiology of parkinsonism
In PD, it is proposed that there is an imbalance of activation of excitatory and inhibitory pathways involving the basal ganglia (a collection of subcortical grey matter nuclei including the substantia nigra, globus pallidus, subthalamic nucleus and the striatum comprising the putamen and caudate nucleus), thalamus and motor cortex governing motor output.3 4 Dopamine usually has an excitatory influence on the direct pathway via D1 receptors to facilitate cortically initiated movements and an inhibitory influence on the indirect pathway via D2 receptors, which usually impedes voluntary movement through thalamic inhibition.3 This pathway is summarised in figure 1.
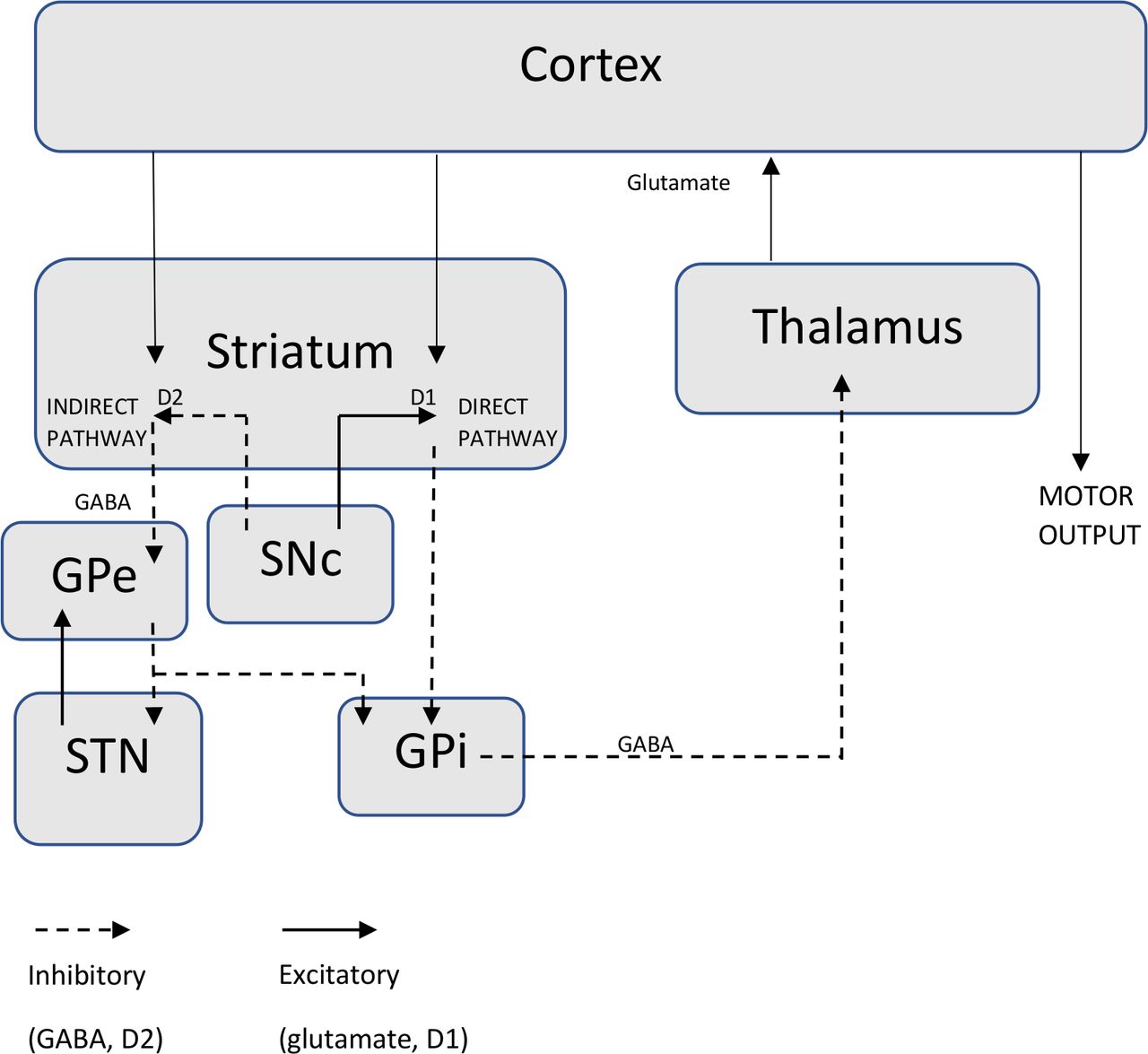
{kind=link}
Schematic diagram of excitatory and inhibitory basal ganglia pathways implicated in parkinsonism. Dopaminergic projections from the SNc have an excitatory influence on direct pathway striatopallidal fibres via D1 receptors leading to disinhibition of thalamic nuclei and enhanced thalamocortical drive facilitating cortically initiated movements. Dopamine also has an inhibitory effect on indirect pathway striatopallidal fibres via D2 receptors, which impedes voluntary movement through thalamic inhibition. The direct pathway is also activated by glutamatergic projections from the sensorimotor cortex and the indirect pathway via gamma-aminobutyric acid (GABA) from the putamen. GPe, globus pallidus externa; GPi, globus pallidus interna; SNc, substantia nigra pars compacta; STN, subthalamic nucleus.
In the case of PD, gradual degeneration of dopaminergic substantia nigra pars compacta neurons and their projections to the striatum leads to motor symptoms.3 A range of dopaminergic medications that either increase dopamine levels through a variety of mechanisms or stimulate dopamine receptors are used to address these symptoms. Conversely, any medication that either blocks dopamine D2 receptors or reduces dopamine release may result in parkinsonism.5 Other aetiologies of parkinsonism typically produce motor symptoms through damage to the basal ganglia, for example, through vascular injury in the case of vascular parkinsonism or normal pressure hydrocephalus.
A degree of parkinsonism is frequent in the elderly and may be due to an age-related decline in the number of nigrostriatal neurons.6 7 However, neurodegenerative pathologies also accumulate with age. A recent study of over 1400 patients followed up for an average 8.5 years until death demonstrated that the majority (94.1%) had at least one pathology on autopsy, the average individual had three pathologies and 25% had PD pathology.8 The other pathologies detected included Alzheimer’s pathology, cerebral amyloid angiopathy, cerebrovascular disease, hippocampal sclerosis and TAR DNA-binding protein 43.8 A majority (53.9%) of this cohort had developed parkinsonism by their last visit.8 Interestingly, this study also highlighted that a proportion of patients with both PD and other pathologies did not demonstrate any clinical parkinsonism.8
Beyond dopamine: the role of other neurotransmitters in parkinsonism and psychosis
Other neurotransmitter systems are also implicated in PD motor symptoms and drug-induced parkinsonism (DIP) and may help explain why DIP may occur with medications that do not have direct effects on dopamine levels or receptors. For example, due to the interrelationship between dopaminergic and cholinergic pathways, parkinsonism has been conceptualised as resulting from relative dopamine deficiency and relative acetylcholine excess.9 This hypothesis is supported by observations that parkinsonism is aggravated by centrally acting cholinergic medications and improved by anticholinergic medications.10 Gait impairment in PD is pathophysiologically complex and freezing of gait, specifically, may be associated with dysfunction in the noradrenergic and cholinergic systems.4
Similarly, contemporary understanding of neurotransmitter systems involved in psychosis has evolved beyond dopamine dysregulation with multiple other neurotransmitters associated with the syndrome. There is evidence that a relative hypocholinergic state favours the emergence of psychotic symptoms in PD.11 PD psychosis has also been conceptualised to reflect an imbalance between dopamine and serotonin neurotransmission.12 Atypical antipsychotics, such as quetiapine and clozapine, demonstrate significant serotonin receptor antagonism.13 This serotonin receptor activity differentiates typical from atypical antipsychotics and was touted as resulting in lower extrapyramidal side effects (EPSE) such as parkinsonism both due to the downstream effect on dopamine release and the need for a lower D2 receptor occupancy for antipsychotic efficacy. With the ongoing use of atypical antipsychotics, it has been demonstrated that there is still a risk of causing EPSE, and the ‘atypical’ nature may be lost with dose increases.
Drug-induced parkinsonism
DIP is defined as the presence of parkinsonism without a history of parkinsonism before the use of the offending drug and onset of parkinsonian symptoms during use of the drug.5 DIP is relevant to patients to a broad range of patients since antipsychotic agents are prescribed for a range of psychiatric indications including adjunctive therapy for depression, anxiety disorders, personality disorders and behavioural and psychological symptoms of dementia.14 This perhaps reflects attitudes that antipsychotic medications are less addictive than, for example, benzodiazepines and may even be perceived as safer. It is possible that patients prescribed these medications for other indications are not properly appraised of the potential risks.14
While antipsychotic medication is a common cause of DIP, other medications have been causally associated with parkinsonism including antiemetic agents,5 antidepressants (selective serotonin reuptake inhibitors),15 lithium16 and calcium channel blocking agents.5
Although DIP is less frequent with atypical antipsychotics than typical agents such as haloperidol,17 it is still commonly seen. A recent Cochrane review concluded that DIP has been associated with all second-generation antipsychotics.18
There is significant individual variability in the propensity to develop parkinsonism in patients treated with causally associated medications which can be only partially explained by reported polymorphisms in genes involved in dopamine transmission in patients who develop DIP,19 or unmasking of subclinical PD or an atypical parkinsonian syndrome.20 Patients with dementia with Lewy bodies (DLB), a closely related synucleinopathy characterised by early cognitive symptoms, are exquisitely sensitive to the effects of dopamine receptor blocking agents.21
Treatment with a dopamine receptor blocker or dopamine depleting agent in a dose or time course consistent with DIP is an exclusion criterion for PD1 and tardive parkinsonism can persist for months or years after cessation of a dopamine receptor blocking agent.22 However, patients who continue to demonstrate parkinsonian features long after causative medications are ceased are thought more likely to have subclinical PD unmasked by neuroleptic exposure.23
Clinical features and differential diagnosis
Differentiating DIP from PD or another underlying neurodegenerative disease is difficult. Both conditions are more frequent with advancing age,5 24 with PD affecting approximately 1% of the population over 60.24 DIP is more common in women5 17 while PD has a slight male predominance.24
Clinical features found to be useful in identifying DIP include a clear temporal association with commencement of potentially responsible medication, bilateral and symmetric features, rigidity, relative absence of rest tremor and the coexistence of tardive dyskinesia.5 25 Freezing of gait is infrequent in DIP compared with other causes of parkinsonism.26 Non-motor symptoms, such as late-onset rapid eye movement sleep behaviour disorder, reflect the involvement of other neurotransmitter systems and are less likely to be a feature of DIP.20 Hyposmia, now part of the supportive diagnostic criteria for PD due to degeneration of the olfactory system,1 can be non-specific, particularly in older patients but is less frequent in DIP.20 Response to levodopa is diagnostically useful with a lack of response or less pronounced response typically seen in DIP.25 Assessing motor response objectively using wearable devices is becoming increasingly useful.27
When parkinsonism develops acutely or progresses rapidly and is associated with other neurological symptoms and signs, other rarer aetiologies should be considered. These include infective and autoimmune encephalitides, structural lesions, stroke and metabolic and genetic disorders.28 Rare secondary causes and investigations can be reviewed in table 1.
Investigations to exclude secondary causes of parkinsonism in atypical cases28 40
Investigations
A number of ancillary investigations have been proposed to differentiate DIP from neurodegenerative pathology. Single-photon emission CT using (123) I-ioflupane as a dopamine transporter ligand (DaT-Scan) is a reliable technique for distinguishing between DIP and PD29 but is not readily available in many countries including Australia. DaT uptake in the striatum is significantly decreased in patients with PD even in early stages of the disease, and drugs causing parkinsonism including antipsychotics have negligible affinity for DaT, so scans may demonstrate normal uptake even with significant DIP.5
Another potentially useful auxiliary investigation is measurement of cardiac sympathetic denervation, an early feature of PD, using metaiodobenzylguanidine scintigraphy,30 but it is not funded as a test for PD in all countries.
MRI and transcranial sonography of the substantia nigra31 32 can demonstrate dopamine depletion with absence of the ‘swallow-tail’ appearance of the substantia nigra and increased iron concentrations, respectively, but the specificity of these tests is uncertain and availability limited.31 32
Finally, a recent pilot study of the utility of cerebrospinal fluid neurofilament light (a biomarker of neuroaxonal damage) in the neuropsychiatric setting found that levels were significantly higher in patients with a neurological or neurodegenerative disorder compared with psychiatric disorder.33 In summary, these tests are promising but still not readily available and clinicians still need to rely largely on clinical acumen.
Management strategies
The treatment of parkinsonism in the psychiatric setting should be individualised, considering varied vulnerabilities and priorities for medication use. Where DIP is a potential cause of parkinsonism, withdrawing or changing potentially responsible medications is the first step.5 DIP usually resolves within months of stopping the offending drug but parkinsonism may persist or progress in 10%–50%.5 While, unmasking of subclinical PD should be considered in this population, parkinsonism has been reported to persist for up to 18 months in some cases of DIP.22
Despite medication withdrawal, patients can be significantly disabled by motor symptoms which may take time to resolve. There is also a risk of unmasking or exacerbating tardive dyskinesia with reduction or cessation of antipsychotics.17 Older and anecdotal evidence exists for the use of anticholinergic agents such as benztropine and levodopa in the symptomatic management of DIP, although it is not robust.25 Anticholinergic medications may also worsen concomitant tardive dyskinesia and amantadine has been proposed as a treatment option in this circumstance.17
Where antipsychotic medication is necessary, lowering the dose or switching to an antipsychotic with a lower risk of DIP such as quetiapine or clozapine is an alternative.5 Quetiapine is relatively safe in patients with parkinsonism and widely used for PD psychosis but has limited evidence for efficacy.34 Clozapine should be used judiciously given the potential for serious haematological and cardiac adverse reactions but, with adequate monitoring, is an effective treatment for psychosis35 with low risk for both parkinsonism and tardive dyskinesia.35 36 The increased mortality associated with antipsychotic use in older patients should also be carefully considered.37
Pimavanserin, a serotonin receptor inverse agonist shown to reduce psychotic symptoms in PD without worsening of motor symptoms,38 is not available for use in all countries.
If psychosis is secondary to a synucleinopathy such as PD or DLB, acetylcholinesterase inhibitors can be considered. Although not adequate for severe psychosis and with potentially limited approved indications, these agents can be antipsychotic sparing and have some evidence in PD psychosis.11
In those with a primary psychiatric disorder, for whom electroconvulsive therapy can be prescribed, it has been shown to improve motor symptoms in both DIP and PD.39
Conclusions
In conclusion, the recognition, diagnosis and treatment of parkinsonism in patients with psychiatric conditions remains challenging. Research on clinical differentiation has aided identification but newer diagnostic investigations are not yet readily available. Underlying neurodegenerative pathology including PD pathology is more frequent in older patients who are therefore more susceptible in the psychiatric context to developing parkinsonism. The reported persistence of symptoms for up to 18 months in cases of DIP highlights the importance of making the diagnosis longitudinally. Cautious changes in medication regimens are required to treat parkinsonism where it arises in the psychiatric setting and new agents are not always necessary.
Acknowledgments
We would like to thank Perminder Sachdev for his assistance in reviewing this manuscript.
References
Footnotes
Contributors AP was involved in the conception of the article, literature review and writing. LG was also involved in the conception and contributed to the literature review and writing. LK critically revised the draft manuscript. MWH was involved in the conception of the review and critically revised the draft manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; internally peer reviewed.