Article Text

Abstract
Background Progressive supranuclear palsy (PSP) is a rare neurodegenerative condition characterised by a range of motor and cognitive symptoms. Very little is known about the longitudinal change in these symptoms over time. Moreover, the effectiveness of clinical scales to detect early changes in PSP is still a matter of debate.
Objective We aimed to determine longitudinal changes in PSP features using multiple closely spaced follow-up time points over a period of 2 years.
Methods
28 healthy control and 28 PSP participants, with average time since onset of symptoms of 1.9 years, were prospectively studied every 3 months for up to 24 months. Changes from baseline scores were calculated at each follow-up time point using multiple clinical scales to identify longitudinal progression of motor and cognitive symptoms.
Results The Montreal Cognitive Assessment, but not the Mini-Mental State Examination, detected cognitive decline at baseline. Both scales revealed poor longitudinal sensitivity to clinical change in global cognitive symptoms. Conversely, the Movement Disorders Society Unified Parkinson’s disease Rating Scale – part III and the PSP Rating Scale (PSPRS) reliably detected motor decline less than 2 years after disease onset. The ‘Gait/Midline’ PSPRS subscore consistently declined over time, with the earliest change being observed 6 months after baseline assessment.
Conclusion While better cognitive screening tools are still needed to monitor cognitive decline in PSP, motor decline is consistently captured by clinical rating scales. These results support the inclusion of multiple follow-up time points in longitudinal studies in the early stages of PSP.
- motor control
- clinical neurology
Data availability statement
Data are available on reasonable request. Data may be made available after reasonable request if appropriate.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Progressive supranuclear palsy (PSP) is a rare neurodegenerative tauopathy and is the most common of the atypical parkinsonian disorders. It is characterised clinically by postural instability with falls, supranuclear vertical gaze palsy, levodopa-unresponsive parkinsonism, dysphagia, dysarthria and cognitive impairment.1 Neuropathological features include neurofibrillary tangles and neuropil threads affecting both neurons and glia in the basal ganglia, brainstem, cerebellum and motor cortex, caused by aggregation of 4-repeat tau. In addition, tau-positive astrocytes contribute to a more accurate diagnosis.2 3
The National Institute of Neurological Disorders and Stroke and the Society for PSP (NINDS-SPSP) first published diagnostic criteria for the classic PSP phenotype, PSP-Richardson Syndrome (PSP-RS), in 1996. However, variability in the PSP spectrum has been discussed for several years,4 including both differing distributions of neuropathology and wide phenotypic divergence. Other PSP variants/phenotypes recognised in post-mortem neuropathological analysis led to the development of new criteria by the International Parkinson and Movement Disorder Society5 (MDS-PSP). The MDS-PSP criteria include different PSP subtypes and show improved sensitivity compared with the original NINDS-SPSP criteria.6 These new criteria define four domains of symptoms, including changes in the oculomotor system, postural instability, akinesia and cognitive dysfunction (cognition, behaviour and speech).
The natural history and clinical features of the disease have been extensively described in the literature.7–10 An RS phenotype, male gender, older onset age, early dysphagia, sleep disturbances, early cognitive impairment and higher PSP Rating Scale (PSPRS) score have been described as predictors of shorter survival.8 11 12
Some well-established and validated clinical instruments have been used in different studies as clinical measures of the progression of symptoms, such as the Unified Parkinson’s Disease Rating Scale13 (UPDRS) and the PSPRS,14 specifically designed for PSP. A task force report15 recommended clinical tools to assess PSP disability and progression, including motor, cognitive, behavioural and functional measures. While these measures have proved to effectively measure clinical symptoms and to report natural history of the disease,7 16 very few studies17 have explored the longitudinal change in different symptom domains. The effectiveness of these clinical scales to detect changes in different PSP features is still a matter of discussion in the literature. While some studies report significant increases in the PSPRS total score over time,18 19 other studies still question its sensitivity to disease progression.20 It is not clear how PSPRS subscales monitor longitudinal changes and which major PSP features decline early in the course of the disease. It has been reported that different PSPRS subscores frequently exhibit ceiling effects.19 Similarly, the MDS-UPDRS motor section (part III) has proved to be efficient assessing motor symptoms in PSP,21 however little is known about how it compares to PSPRS in its ability to monitor disease progression. Delays in the establishment of a diagnosis may explain the well described rapid progression of the disease but also ceiling effects seen in some PSPRS domains that may start to decline earlier in the course of the disease. Thus, it is of utmost importance to prove the clinical validity of this tool in earlier stages of PSP and to detect early changes in symptoms in individuals with a shorter disease duration.
Cognitive impairment is a key feature in PSP diagnosis with patients showing a vast range of deficits in different cognitive domains such as executive functions, verbal fluency, attention and visuospatial functions (for a review see22). A comprehensive assessment of the full neuropsychological profile of PSP patients is not always possible to perform, both in research and clinical settings. Brief cognitive screening tests such as the Mini-Mental State Examination23 (MMSE) and the Montreal Cognitive Assessment24 (MoCA) are useful tools to support the diagnosis of neurodegenerative diseases. These scales were not designed specifically for PSP and their sensitivity to detect cognitive decline in this condition is still a matter of discussion in the literature.25
Overall, it is fair to say that the sensitivity of the existing clinical measures to detect changes in PSP trajectory over time is still a matter of debate. Moreover, most of the recommended clinical tools were validated in populations of patients several years after symptom onset and it is still not clear how well they monitor changes in symptoms in the early stages of the disease. In this study, we aimed to determine longitudinal changes in symptoms of PSP using different clinical measures at multiple closely spaced follow-up time points.
Methods
Participants
The participants were recruited as part of the Oxford Quantification in Parkinsonism (OxQUIP) study (demographics summarised in table 1; further information about the study described in online supplemental table 1).
Supplemental material
Demographics and baseline (visit 1) scores for HC and PSP participants
All PSP participants received a diagnosis of possible or probable PSP, according to the MDS PSP criteria5 by a consultant neurologist. Healthy control (HC) individuals were spouses/caregivers of the OxQUIP participants and had no history of neurological or psychiatric disorders at the time of testing. All participants were given written information about the study, and informed consent was obtained prior to participating.
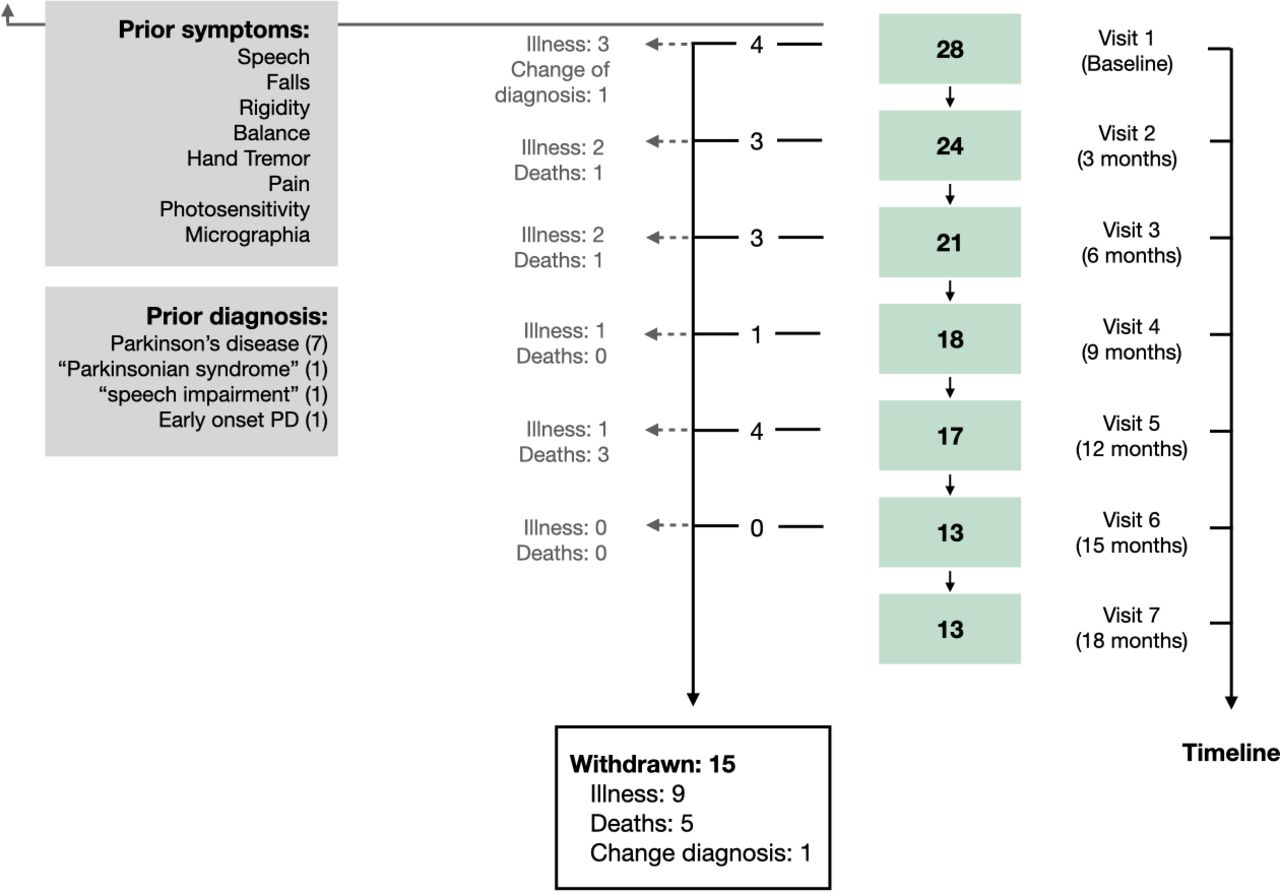
Each visit lasted no more than 2 hours and it comprised two parts: motor and cognitive assessment. Nine visits were planned over 24 months, one baseline visit followed by eight visits at months 3, 6, 9, 12, 15, 18, 21 and 24 (figure 1). However, only data from the first seven visits (ie, the first 18 months of follow-up) are analysed here, due to the fact that the already high dropout rate was compounded at visits 8 and 9 by the discontinuation of some participants due to COVID-19 lockdown restrictions (all visit seven time points had passed prior to the pandemic).
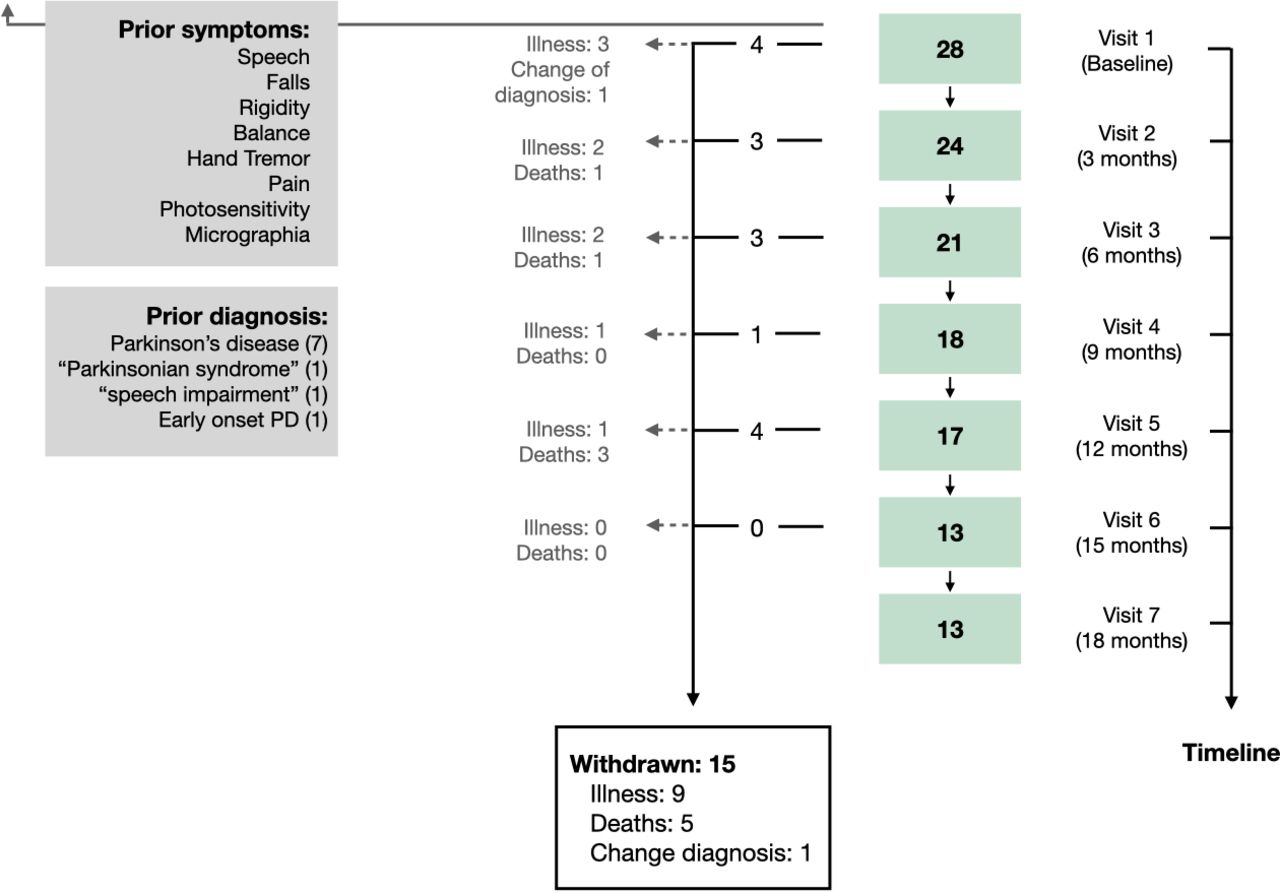
Flow chart illustrating the inclusion and lost to follow-up of participants at each visit time point. Twenty-eight cases with progressive supranuclear palsy were recruited in 2016–2020. Participants completed a baseline assessment at visit one and underwent a follow-up assessment every 3 months over a period of 18 months. Of the 28 participants enrolled in the OxQUIP study, 15 withdrew due to a combination of reasons including: being too unwell to attend visits (9), death (5) or to a change of diagnosis (1). Participants reported as early clinical PSP-related symptoms prior to the first OXQUIP visit the following symptoms: speech impairments, falls, rigidity, problems with balance, hand tremors, pain, photosensitivity and micrographia. The figure also shows previous diagnosis received by the participants prior to the PSP diagnosis. PSP, progressive supranuclear palsy; PD, Parkinson’s disease; OxQUIP, Oxford Quantification in Parkinsonism.
Research protocol
Demographic data were collected according to the OxQUIP protocol26 and the following information was initially collected at baseline assessment: time since onset of symptoms, time since diagnosis, age at onset of symptoms, laterality, previous diagnosis, early and predominant symptoms and medication details. Time since onset of symptoms was defined as the time since the earliest reported clinical symptom attributable to a parkinsonian diagnosis, to their baseline visit. Time since diagnosis was considered as the time elapsed from PSP diagnosis to baseline assessment. For all subjects, the month and year of disease onset was available, and the exact date was taken as the first day of the month for statistical analysis purposes.
Motor and cognitive assessment was performed using the OxQUIP study protocol, that includes the Motor section (part III) of the MDS—UPDRS,13 the Hoehn and Yahr stage of illness,27 the PSPRS,14 the MMSE,23 the MoCA,24 the verbal fluency test of the Delis–Kaplan Executive Function System Test28 and the Schwab and England Activities of Daily Living Scale.29
Statistical analysis
All statistical analyses were performed using SPSS V.25.0 (SPSS).
Normality of distributions was checked with the Shapiro-Wilk test, and homogeneity of variance with Levene’s test. Normally distributed data for PSP and control participants were compared with a t-test (independent samples) and non-normally distributed data with a Mann-Whitney U test. Categorical data were compared with a χ2 test.
Changes from baseline to each visit in the different motor and cognitive scales were analysed with a paired t-test and a Wilcoxon signed-rank test, for normal and non-normally distributed data, respectively. Delta (∆) scores for each measurement were defined as the total/subtotal score of a scale at each visit minus baseline visit scores. The absolute value of Cohen’s d and its correspondent non-parametric value ( ) were calculated for each change in clinical measure over time as measures of effect size. The association between clinical and demographic variables was explored using Pearson R2 and Spearman correlation coefficients where appropriate.
) were calculated for each change in clinical measure over time as measures of effect size. The association between clinical and demographic variables was explored using Pearson R2 and Spearman correlation coefficients where appropriate.
Baseline and clinical variables between completers and those who withdrew from the study were compared with a t-test (independent samples), or with a Mann-Whitney U test for non-normally distributed data. To investigate the effects of baseline variables in the dropout rate among PSP participants, a bivariate logistic regression was performed.
Post hoc analyses, with Bonferroni adjustment for multiple comparisons, were used when appropriate. All p values less than 0.05 were considered statistically significant.
Results
Demographics and clinic visits
Twenty-eight PSP and 28 HC participants were recruited. Demographics and baseline scores for HC and PSP participants are summarised in table 1. Participants with PSP had fewer years of formal education (p=0.013) than HC but there were no differences in terms of age or gender. As expected, PSP participants were more impaired both in the motor and the neuropsychological assessments than HC (p<0.01, see table 1).
Fifteen (54%) of the PSP participants had withdrawn from the study prior to the end of the analysis period (visit 7 at 18 months): nine due to being too unwell to attend study visits, five due to death and one due to change in diagnosis (this patient was excluded from the analysis). Although the number of participants decreased at each visit, only 3 (11%) subjects attended one single visit and 12 (43%) made more than five visits, all at a 3-month interval. Mean time from onset of symptoms to the first visit was 1.9 years (range 0.2–6.3) and mean time since PSP diagnosis to first visit was 1.2 years (range 0.1–5.10).
Mean changes from baseline assessment of clinical measures
Figure 2 show the absolute values of PSPRS and MDS-UPDRS-III for PSP participants at each of the seven visits. Horizontal bars represent means. Figure 2 show changes in the same variables with respect to baseline values. Asterisks represent group level differences from baseline that are significant at the p<0.05 level (*) and the p<0.008 level (**), the latter incorporating a Bonferroni correction for multiple comparisons.
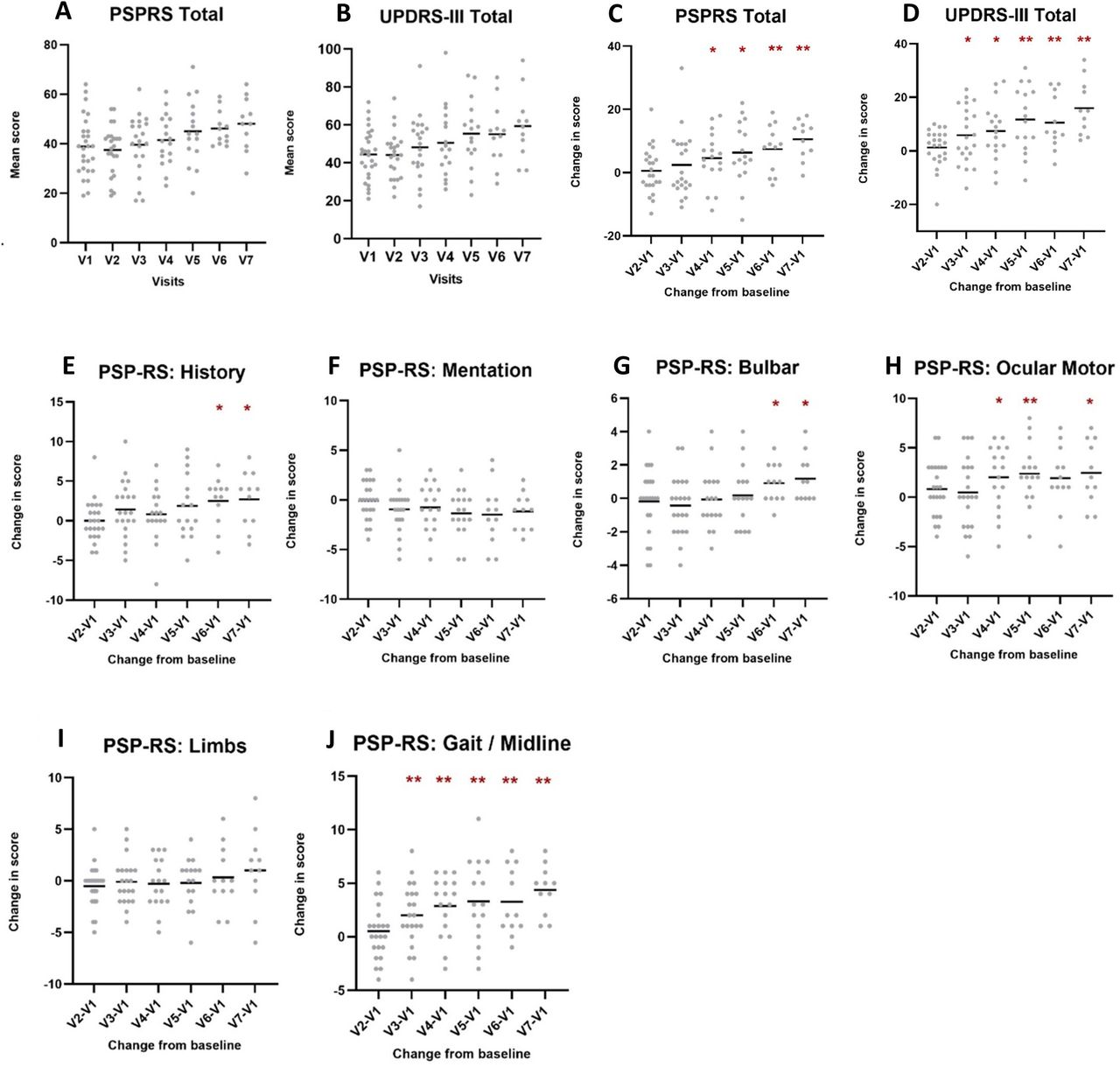
Mean scores at each visit (A, B) and mean changes from baseline (C, D) in MDS-UPDRS-III and PSPRS total scores. Mean changes from baseline in PSPRS subscores (E–J). To determine the mean change in scores from baseline, ∆ values were calculated for each participant and scores were averaged for each visit. Figure shows values significant at level *p<0.05 and **p<0.008, the latter including adjustment for multiple comparisons with a Bonferroni correction. PSPRS, Progressive Supranuclear Palsy Rating Scale; MDS-UPDRS-III, Movement Disorders Society Unified Parkinson’s disease Rating Scale part III.
As a measure of their suitability as longitudinal progression markers, we were interested in determining how long it takes for each variable to show a statistically significant shift from baseline values at the group level. Significant variations from baseline at single time points may represent statistical fluctuations and so we defined an enduring change as having occurred when scores were significantly different to baseline results for at least two consecutive visits.
The MoCA and MMSE scores did not show any enduring changes in scores over time. Conversely, PSPRS and MDS-UPDRS III total scores both showed enduring changes over time (figure 2C,D). The increase in MDS-UPDRS III was statistically significant after 12 months (∆=11.75, SD=12.31, p<0.008; figure 2D) while the increase in PSPRS became significant 3 months later, 15 months after baseline assessment (∆=7.42, SD=7.63, p<0.008; figure 2C).
When looking at groups of symptoms, it becomes evident that PSPRS subdomains have different patterns of progression. As shown by figure 2E–J, the ‘Gait / Midline’ and ‘Ocular Motor’, subsections show the earliest enduring changes over time, after 6 (∆=2.00, SD=4.92, p<0.008) and 9 months (∆=2.38, SD=3.01, p<0.008), respectively. This is followed by ‘Bulbar’, and ‘History’ at 15 months (‘Bulbar’: ∆15 months=1.06, SD=7.02, p<0.05; ‘History’: ∆15 months=2.50, SD=3.09, p<0.05). ‘Mentation’ and ‘Limbs’ subsections did not show an enduring change from baseline during the 18-month follow-up in this study.
Table 2 shows ∆ scores, SD and effect sizes for the earliest significant change from baseline scores (total and subtotal scores) in each clinical measure at p values of both <0.05 and<0.008 (after Bonferroni correction). As expected, MDS-UPDRS-III total score was positively correlated (p<0.01) with PSPRS total score at most visits.
Enduring changes in scores in clinical measures with and without adjustment for multiple comparisons
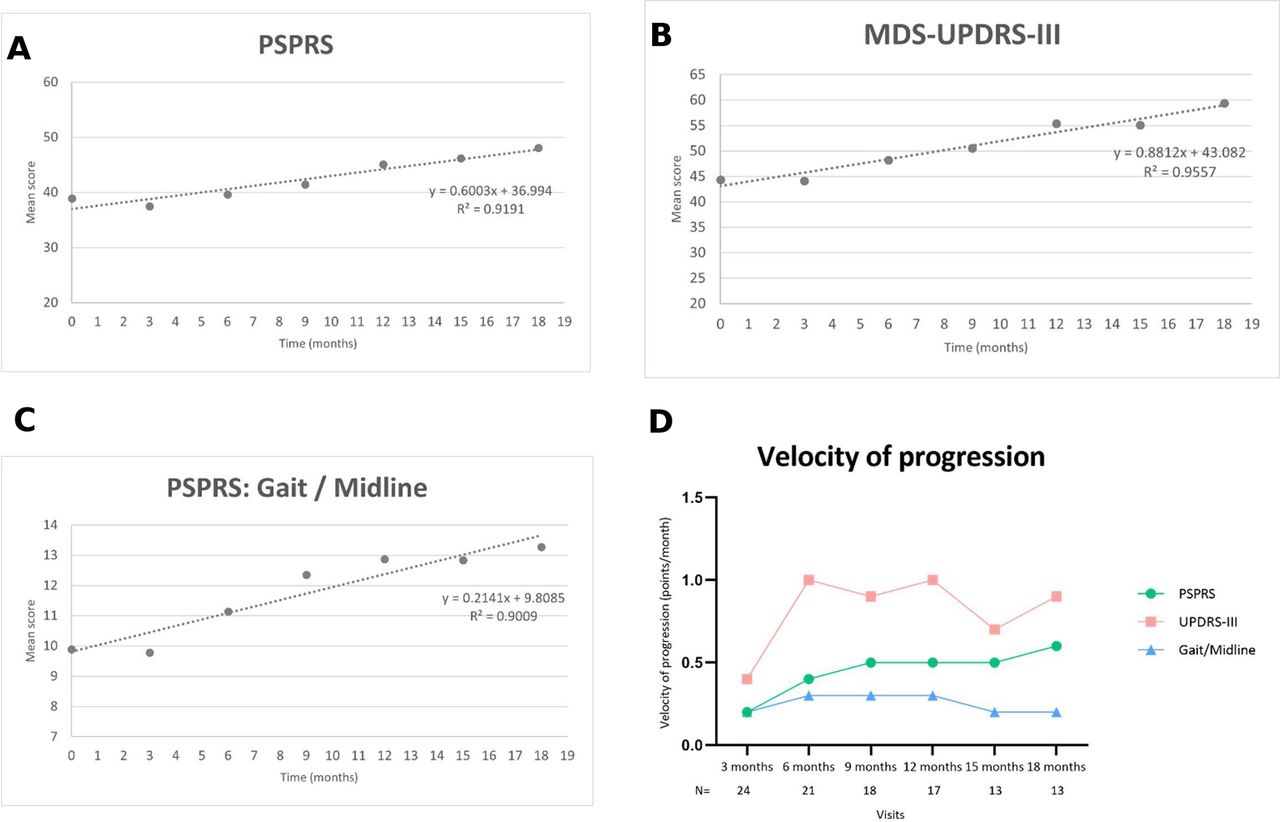
The three scores exhibiting the earliest and most consistent changes over time were MDS-UPDRS-III, PSPRS and the PSPRS gait/midline subscore. For each of these, the group means at each visit were plotted against time and fitted with a straight line (figure 3). The gradient of the line provides an estimate of the rate of change of the marker with time. MDS-UPDRS-III was found to progress at 0.88 points per month (8.0% of maximum score per annum, R2=0.96), PSPRS at 0.60 points per month (7.2% of maximum score per annum, R2=0.92), and PSPRS gait/midline subscore at 0.21 points per month.
{kind=link}
{kind=link}
{kind=link}
Velocity of progression of the enduring changes in total scores of PSPRS (A), MDS-UPDRS-III (B) and PSPRS ‘Gait/Midline’ subscore (C). Figure D shows a comparison between the velocity of progression of the enduring changes in scores represented in figures A–C. N=number of participants at each visit. MDS-UPDRS-III, Movement Disorders Society Unified Parkinson’s disease Rating Scale part III.; PSPRS, Progressive Supranuclear Palsy Rating Scale
Withdrawal rate
All comparisons between baseline scores and each of the follow-up scores considered only participants who completed both assessments. In order to detect some possible biases introduced by the high withdrawal rate, we first compared baseline demographic and clinical characteristics between completers and those who dropped out of the study (online supplemental figure 1). No significant differences were found for education, time since diagnosis and time since onset of symptoms between both groups. However, participants who dropped out were older at first visit (t(25) = −4.237, p<0.001) and older at onset of symptoms (t(24) = −3.819, p=0.001) than completers. Given the non-significant trend to improvement in the PSPRS mentation subscore, this score was also compared at baseline and at each visit between completers and those who dropped out from the study; no significant differences were found at any point. No differences were found for any other clinical measures between both groups, after adjusting for multiple comparisons (Bonferroni correction p<0.01).
A binary logistic regression analysis (with ‘backwards’ method) was then performed to assess if any of the baseline demographic and clinical characteristics could be a potential predictor of the drop-out rate. We found that participants with an older age at onset of symptoms were more likely to withdraw (online supplemental table 2).
Discussion
The aim of this study was to examine at short timescales the progression of PSP symptoms, using established clinical measures. We performed a longitudinal analysis over an 18-month period with visits at 3 monthly intervals. Clinical measures captured a steady decline of many motor symptoms in this short time. Cognitive screening measures detected cognitive decline at baseline, but did not demonstrate progression of cognitive decline over the study period.
PSP participants had some degree of cognitive impairment at baseline and were classified differently using the two standard cognitive measures. While they scored within the normal range in the MMSE (M=26.0, range=20–30), MoCA scores fell below the cut-off score of 26 (M=22.4, range=12–30) at baseline. This discrepancy mirrors previous findings reporting a better sensitivity of the MoCA in detecting global cognitive decline in other pathologies such as Alzheimer’s disease (for a review see ref. 30) and Parkinson’s disease,31 and also in PSP.25 The overall ceiling effect observed in most of the subitems in the MMSE and the fact that the MoCA is more sensitive to executive functioning and attention performance than the MMSE,25 32 makes the MoCA a more suitable tool to detect mild global cognitive impairment in PSP. Our results corroborate that early cognitive impairment is a key feature in PSP and add new evidence that this impairment can be found even in individuals with a short disease duration. Despite the ability to detect some degree of cognitive impairment at baseline, these global screening tests lack domain-specificity and fail to monitor progression of the well-established frontoexecutive deficits commonly found in PSP.33 A more detailed assessment of the progression of executive functions in early stages of PSP is needed in order to track cognitive changes in PSP longitudinally.
While we were not able to detect progression of cognitive symptoms using standard clinical tools, several motor symptoms were clearly shown to decline over the study period. We found that PSPRS and MDS-UPDRS-III have good sensitivity to longitudinal change in motor symptoms, as demonstrated by the comparable effect sizes in both scales. Although the MDS-UPDRS was not specifically designed to monitor PSP symptoms, changes in motor scores mirror those of the PSPRS. As well as detecting motor deficits in PSP,21 MDS-UPDRS part III also appears suitable to monitor disease progression. Our study supports previous findings19 about the sensitivity of PSPRS total score as a measure of clinical progression in PSP, and adds evidence that MDS-UPDRS-III is also a good measure of motor progression. In this study group, MDS-UPDRS-III actually detected progression slightly earlier than PSPRS, and we would therefore advocate its routine use alongside PSPRS in PSP trials.
We found a relatively slow yearly progression in the PSPRS total score (6.3 points/year; 7.2%) compared with those found in previous studies,14 34 35 (see table 3). In comparison to previous studies where the average disease duration at first assessment was approximately 3 years,7 14 the present sample was recently diagnosed and the average time since the onset of symptoms was 1.9 years. With a 1.5-year study period, the average participant will have completed our study when they would have only just entered the others. This raises the possibility that an accelerating progression pattern could account for at least some of the discrepancy in progression rates between studies. Figure 3D may be suggestive of acceleration in the change in PSPRS which would support this hypothesis.
Changes (mean, SD) from baseline to follow-up at 12 months: OxQUIP versus other PSP studies
PSPRS subscores revealed different patterns of progression with time. ‘Gait/Midline’ and ‘Ocular motor’ showed the earliest changes, and ‘History’ and ‘Bulbar’ had a later decline. These changes are very much in line with the new MDS-PSP diagnosis criteria5 proposing ocular motor dysfunction, postural instability and akinesia as core motor features of the disease. The ‘Gait/Midline’ subscore had the earliest change in score observed at 6 months, as reported before.36 Moreover, this decline was consistent at each follow-up time point after the first significant change in score 6 months after baseline. These enduring changes in ‘Gait/Midline’ indicate that this measure could be used in short interval clinical assessments and in future clinical trials, as previously suggested by other studies.37 Moreover, our study further extends these findings by detecting this early decline in patients with a disease duration less than 3 years. Our earlier work has shown that using digital technology to measure gait accurately can yield an effective diagnostic tool for discriminating PSP from PD.38 It is, therefore, possible that this same marker could be used for both diagnosis and disease progression monitoring.
‘Bulbar’ and ‘History’ PSPRS subscores had a later decline, with an enduring change in score only emerging after 15 months, and in both cases this did not reach the Bonferroni-corrected significance level. This is consistent with previous studies showing that dysphagia and dysarthria decline in later stages of the disease.39 Interestingly, no significant change over time occurred in the ‘Mentation’ subscore, showing that this measure does not capture disease progression of specific cognitive-behavioural/emotional symptoms at an early stage and it might be particularly prone to inter-rater variability,14 36 nor was there significant change in the ‘Limbs’ subscore.
Limitations
Our study has some limitations that are important to consider. First, the sample size is relatively small. Despite this, and notwithstanding the high withdrawal rate due to disease progression, the results obtained during the first 18 months of the study had good statistical power even after adjusting for multiple comparisons, with medium to large effect sizes. High drop-out rates are common in PSP studies due the rapidly progressive nature of the condition, and in an effort to identify any potential bias caused by the high dropout rates, we compared baseline characteristics between those who dropped out and those who completed the study. No differences were found on clinical measures and on most demographic variables in both groups, suggesting that these characteristics do not have a meaningful impact on study completion. However, being older at the onset of symptoms slightly increased the likelihood of withdrawing from the study. Despite this, a potential impact of the high withdrawal rate on the study outcome remains possible. As discussed previously in the literature (Williams-Grey et al), the main methodological issue in longitudinal studies of this type is a potential for drop out bias. Withdrawal can potentially introduce bias if those lost to follow-up differ clinically from those who continue to be assessed. It may be that those dropping out would on average have been found to be progressing more rapidly on the measures assessed than those who stayed in the study. This leads to the potential for underestimation of progression rates. This is a factor to be considered when designing clinical trials. Many of our participants travelled considerable distances to the single study centre, and this will likely have played a role in their decision that they could no longer travel to the test centre as their illness progressed. Possible mitigations for future studies include the use of more test centres to shorten travel distances, or even remote testing in the participant’s home.
Another important limitation is that the PSP diagnosis was clinical rather than pathological. Seventeen participants were subsequently classified as PSP-RS phenotype and 10 as PSP-Parkinsonism. To date, seven participants have died, four of whom had a postmortem pathological confirmation of diagnosis. Earlier studies suggest PSP-RS has a faster progression than other phenotypes40 but the numbers in this study were not sufficient to explore phenotypic differences.
Some of the participants were on antiparkinsonian medication, and this was not stopped for study visits. Dopaminergic medication has a well-documented confounding effect on motor assessment in studies of Parkinson’s disease. The response to medication in PSP is relatively weak, but nonetheless we cannot exclude that this may have reduced apparent progression rate to a degree.
In conclusion, determining the earliest changes in symptoms is a crucial step for the success of therapeutic trials in PSP, particularly when one considers the short prognosis of this condition. To the best of our knowledge, our study is the first describing the progression of PSP clinical symptoms in the early stages of the disease with such closely spaced follow-up time points. It was possible to determine enduring changes in certain domains as early as 6 months into the study. This supports the inclusion of multiple closely spaced follow-up assessments in future clinical trial protocols.38 41
Data availability statement
Data are available on reasonable request. Data may be made available after reasonable request if appropriate.
Ethics statements
Patient consent for publication
Ethics approval
The measurements were obtained during the Oxford study of QUantification In Parkinsonism (OxQUIP), a longitudinal study conducted at the John Radcliffe Hospital in Oxford, UK. The study was approved by the research ethics committee and the Health Research Authority (REC 16/SW/0262).
Acknowledgments
We thank the participants and their families for their endless support with our research work.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @NeuroMetrology
Contributors We would like to thank the patients and their families for their tremendous support with OxQUIP. CAA is responsible for the overall content as guarantor. CAA and JJF conceived this article. MFP, CAA and JJF wrote the manuscript with further contributions from TB, GUH, MB, GT, SP and NS. All the authors contributed to critical revisions and approved the final version of the article. CAA accepts full responsibility for the finished work and the conduct of the study and has access to the data presented in this study.
Funding Grant from UCB Biopharma SRL. JJF was supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.