Article Text

Abstract
Objective To evaluate the interim feasibility, safety and clinical measures data of direct delivery of regenerating peripheral nerve tissue (PNT) to the substantia nigra (SN) in participants with Parkinson’s disease (PD).
Methods Eighteen (13 men/5 women) participants were unilaterally implanted with PNT to the SN, contralateral to the most affected side during the same surgery they were receiving deep brain stimulation (DBS) surgery. Autologous PNT was collected from the sural nerve. Participants were followed for safety and clinical outcomes for 2 years (including off-state Unified Parkinson’s Disease Rating Scale (UPDRS) Part III assessments) with study visits every 6 months.
Results All 18 participants scheduled to receive PNT implantation received targeted delivery to the SN in addition to their DBS. All subjects were discharged the following day except for two: post-op day 2; post-op day 3. The most common study-related adverse events were hypoaesthesia and hyperaesthesias to the lateral aspect of the foot and ankle of the biopsied nerve (6 of 18 participants experienced). Clinical measures did not identify any hastening of PD measures providing evidence of safety and tolerability. Off-state UPDRS Part III mean difference scores were reduced at 12 months compared with baseline (difference=−8.1, 95% CI −2.4 to −13.9 points, p=0.005). No complications involving dyskinesias were observed.
Conclusions Targeting the SN for direct delivery of PNT was feasible with no serious adverse events related to the study intervention. Interim clinical outcomes show promising results meriting continued examination of this investigational approach.
Trial registration number NCT02369003.
- PARKINSON'S DISEASE
- NEUROSURGERY
- SCHWANN CELL
Data availability statement
No data are available.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic
The loss of functional neurons in the substantia nigra is a hallmark of Parkinson’s disease. Finding a way to stabilise or slow the neurodegeneration of these neurons continues to be an ongoing effort.
What this study adds
The procedure of deploying autologous reparative peripheral nerve tissue to the substantia nigra is feasible and has not led to serious adverse events related to the implantation.
How this study might affect research, practice or policy
While unilateral delivery appears to be a feasible and safe investigational procedure, expanding the scope of deployments to bilateral substantia nigra deliveries may help in determining the appropriate amount of reparative peripheral nerve tissue to use for future trials.
Introduction
No disease modifying therapies currently exist for Parkinson’s disease (PD). Investigational biologic or cell therapies for treating neurological disorders typically require the direct delivery of a novel agent into a specific anatomic target or targets within the central nervous system. Many clinical trials focusing on PD have used a direct delivery method to evaluate the potential of growth factors,1–5 gene therapies,6–8 fetal brain tissue9 10 or stem cells11 for their ability to have a potential impact on therapeutic outcomes. Direct delivery methods are associated with surgical risks and present organisational, procedural and ethical challenges for the participants and research teams, especially when performed first in human, stand-alone procedures in preliminary clinical trials.12 The substantial need for disease altering therapies for PD is the major driving force for this line of work.13
Our approach focuses on developing a potential disease modifying intervention based on cell therapeutic principles. The potential mechanism of action of cell-based therapies is postulated to be either a triggering event, where the implanted cells do not survive, but trigger a lasting reaction within the host brain, or a living therapy, where the cells survive long term and perform a designated function to influence the host environment. Living therapies for neurodegenerative disorders can involve cell replacement strategies, such as the transplantation of fetal dopaminergic neurons,9 10 or cellular support strategies that facilitate repair and neuroprotection.1 2 7 14 For our approach, we chose to use autologous peripheral nerve tissue (PNT) as a strategy to provide cellular support to the diseased and dying neurons within the region of the substantia nigra (SN). Peripheral nerves demonstrate a robust ability to repair themselves following injury through the production of many cell-growth and cell-survival factors, as well as cell-to-cell interactions, leading to axonal regeneration and return of function (reviewed in 15 16).
Our surgical protocol design integrates the direct delivery of PNT with deep brain stimulation (DBS), a standard of care, US Food and Drug Administration (FDA)-approved, surgical approach to the symptomatic treatment of advanced PD.17–19 This integrated platform, which we have termed DBS-Plus, offers multiple advantages, and some associated limitations and challenges, which are addressed in this report.
The major goals of this study were to evaluate the safety of the DBS-Plus surgical approach and the potential consequences of the direct delivery of a cell therapy intended to integrate into the delivery area and survive for a long period of time.
We have previously reported our proof-of-concept results20 and corresponding feasibility and 1 year safety data21 in eight participants trialling PNT implantation to the SN and DBS targeting to the subthalamic nucleus (STN). The current report builds on our previous experience and provides feasibility, safety and clinical outcome data from 18 new participants trialling PNT implantation to the SN and DBS primarily targeting the globus pallidus interna (GPi). The results provide a midpoint interim evaluation from the original trial designed with a 2-year study duration. Complete results, including imaging, neurocognitive and clinical outcomes, will be reported after completion of the 2-year trial by all subjects.
Methods
Study design
Investigator initiated, open-label, single-centre, phase I trial focusing on the feasibility and safety of the direct delivery of PNT. Patients deemed to be qualified candidates for DBS and who had agreed to receive DBS were informed about the study and asked to participate (figure 1 and box 1). The trial design also includes reporting of clinical evaluations and outcome data to assist in the determination of those measures that may prove clinically meaningful for evaluating disease progression and the potential for disease modification.
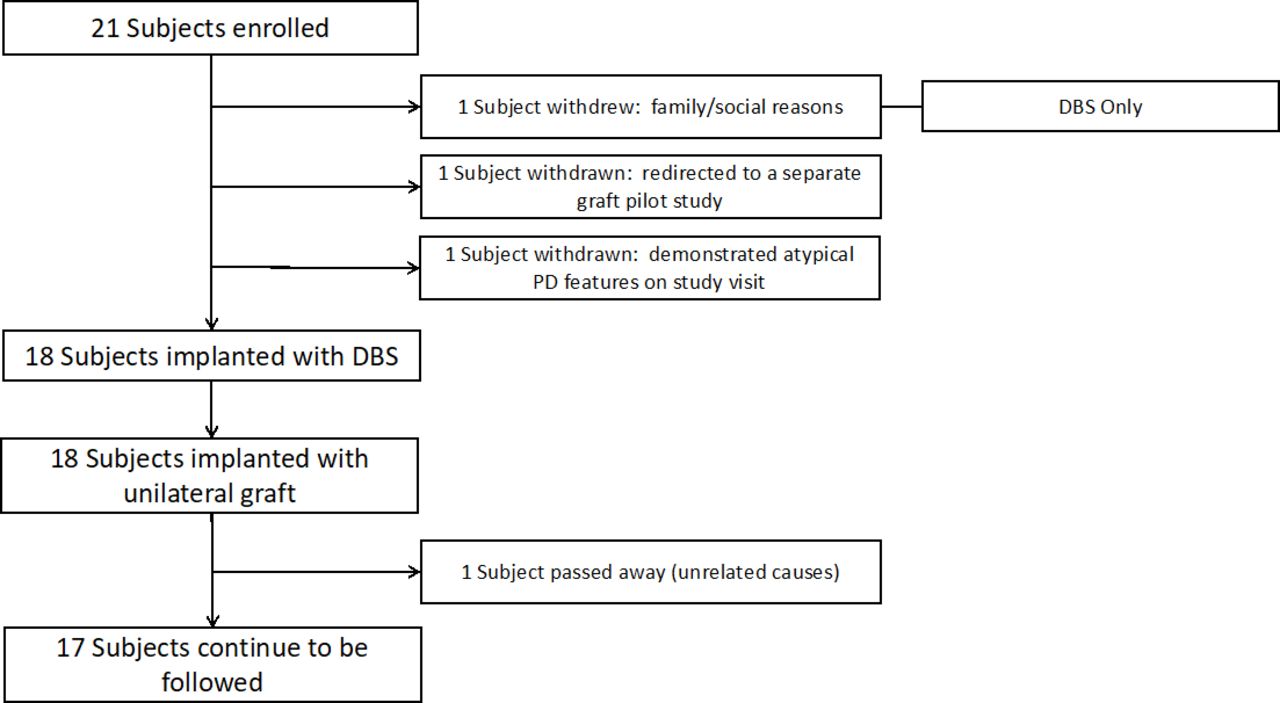
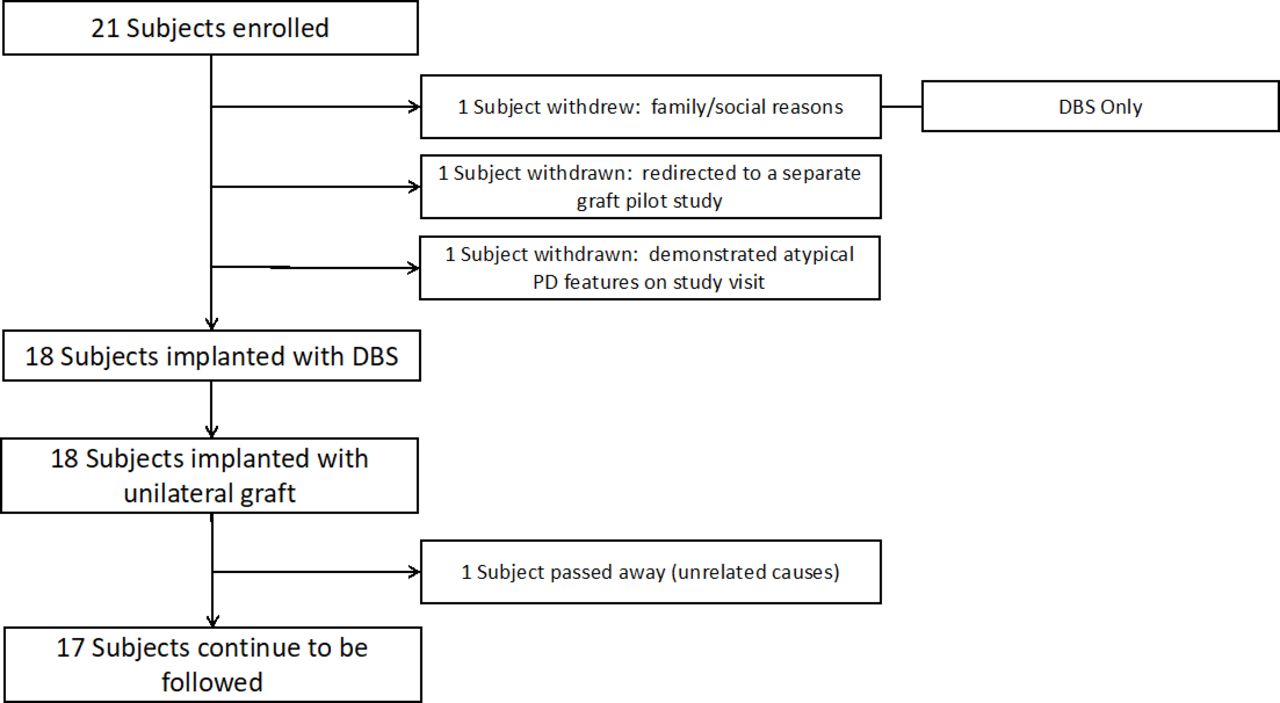
Enrolment and surgical flow. DBS, deep brain stimulation; PD, Parkinson’s disease.
Inclusion and exclusion criteria
Inclusion criteria
Undergoing deep brain stimulation (DBS) of the subthalamic nucleus or globus pallidus interna
Between the ages of 40–75
Able to give informed consent
Show a positive response to Levodopa
Show no significant cognitive deficit per a formal neurocognitive exam
Be able to tolerate the surgical procedure
Exclusion criteria
Any condition that would make the subject a poor candidate for DBS of any target
Under the age of 40 or over the age of 75
Unable to give informed consent
Previous Parkinson’s disease surgery or intracranial surgery
Study approvals and monitoring
The University of Kentucky Institutional Review Board approved the study, and data from the trial were regularly reviewed every 4 months by the Center for Clinical and Translation Science Data and Safety Monitoring Board at the University of Kentucky. All participants provided written informed consent. The trial protocol was developed and was carried out in accordance with the FDA’s Same Surgical Procedure Exemption, 21 CFR 1271.15(b). Go/nogo requirements were based on adverse event (AE) reporting and adjudication. If three or more AEs were considered to be severe, and related to the study, then the study would have to be halted until further investigation and evaluation.
Imaging
MRI
Preoperative MRIs (3T) were obtained before surgery for targeting purposes. Thin cut CT images were obtained following Cosman-Roberts-Wells (CRW, Integra, Princeton, New Jersey, USA) frame placement and then fused to the preoperative MRIs for stereotactic planning. Postoperative MRIs (1.5T) were obtained within 36 hours of the implantation surgery during the participant’s inpatient stay. MRIs were evaluated clinically by neuroradiology faculty.
Single photon emission computed tomography (SPECT)
All participants underwent 123I-ioflupane SPECT scans at two time points, preoperative and 24 months postoperative.
Image acquisition
Four hours after injection of 5.6 (0.3) mCi, Mean (SD), of 123I-ioflupane (DaTscan), SPECT images of the head were acquired using a gamma camera (Symbia, Siemens, USA).
Image analysis
Both qualitative and quantitative analyses of the basal ganglia comparing preoperative and 24-month postoperative timepoints were performed. For quantitative analysis, specific binding ratio (SBR) was calculated for the left and right striata, using DaTQUANT software (V.2.0, GE Healthcare, Boston, Massachusetts, USA), according to the formula: SBR = ((region count density)/(occipital region count density))− 1. The preoperative evaluations are presented here, and the 24-month comparisons will be reported at the conclusion of the study. Consensus between the qualitative assessment and identification of a normal striatal signal pattern serves as an exclusionary criterion for the clinical diagnosis of PD.22
Surgical implantation
The DBS-Plus protocol is designed to allow the direct delivery of an experimental agent within the same operative setting as DBS without interfering with the DBS procedure. In the current trial, PNT was implanted after the bilateral DBS electrodes had been placed, tested, secured to the skull and connected to the lead extensions. The specific details have been previously described.20 23
In brief, all stereotactic procedures were performed under general anaesthesia using a CRW frame. DBS and PNT trajectories were planned individually using stereotactic planning software (IPlan V.3.0 Stereotaxy, BrainLab, Munich) to enter through a cortical gyrus in the region of the coronal suture and lateral enough to avoid the lateral ventricles. Participants received bilateral DBS electrodes: 17 to the GPi and 1 to the STN. Microelectrode recordings were used to aid in target localisation. All trajectories were targeted to avoid intraparenchymal vasculature visible on MPRAGE sequences. Entry zones for the PNT implant cannula were lateral to the DBS electrode entry zone and traversed the same burr hole as the electrode. Implant cannula was simply a standard guide tube with a 5-mm long side window.20
PNT was implanted unilaterally into the SN contralateral to the most affected side based on Unified Parkinson’s Disease Rating Scale (UPDRS) Part III scores. The centre portion of the SN, based on susceptibility weighted imaging (SWI) hypodensity, was used for targeting the centre of the PNT deployment. Implant cannula capacities were 5 mm in length with a 1.5 mm diameter. The PNT consisted of five to ten 1 mm segments cut from fascicles harvested and dissected from the distal component sural nerve that had been transected during the first stage of the multistage DBS surgery.23 In accordance with the FDA’s Same Surgical Procedure Exemption, PNTs were not exposed to any chemical or biological elements beyond sterile saline, and the only manipulations were dissection, resizing and reshaping. Immunosuppressants were not used because the PNT is autologous tissue.
Analysis of implant cannula trajectory and implant placement
Implant cannula trajectories were easily identified on postoperative MRI MPRAGE and SWI sequences. Implant locations were also identified and designated by placing an object marker within the mid portion of the implant delivery zone (2.5 mm from the distal terminus). Accuracy of implant targeting was determined by localising the implant marker in relationship to the SN as identified both visually and with the anatomical 3D overlay within Elements (Brainlab, Munich). Because the SN lies within a plane orthogonal to the Euclidian planes designated by the anterior commissure-posterior commissure line (AC-PC) line and the coronal plane, the figures generated for use in depicting the implant sites were oriented along the long axes of the SN (figure 2).
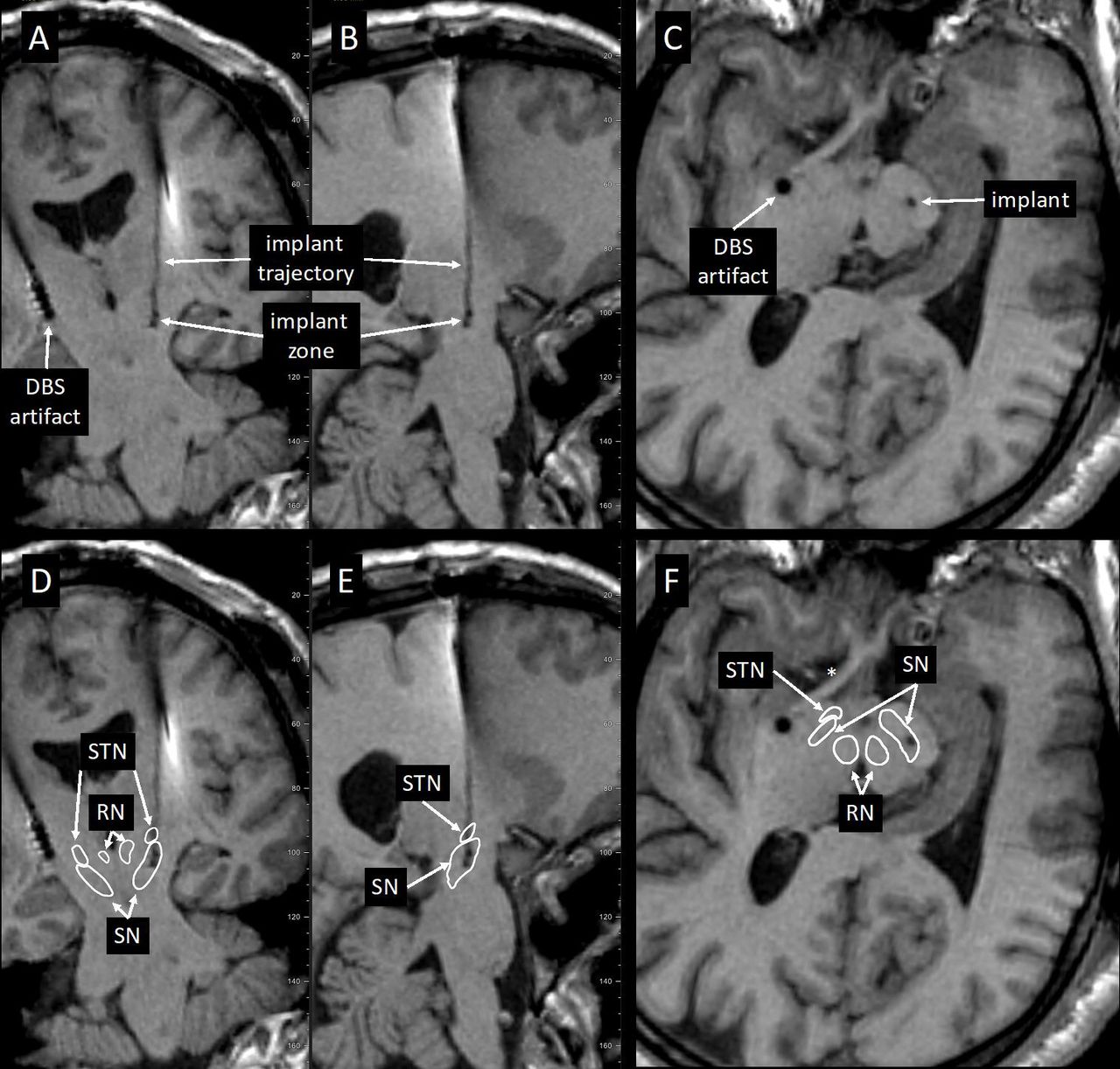
Postoperative MPRAGE scans displaying deep brain stimulation (DBS) electrode and graft cannula (implant trajectory and zone) tracks. DBS electrode and graft cannula placement in the coronal (A), parasagittal (B) and orthogonal to the trajectory (C) planes. Outlines of the substantia nigra (SN), subthalamic nucleus (STN) and red nucleus (RN) superimposed on the images (D–F) show implant zone relative to the SN.
Safety and feasibility
Safety data for each participant were collected starting from the first stage of surgery to the end of the study. Feasibility data were collected for each participant from the time of enrolment to the end of the study and were broken down into procedural and compliance data. Procedural feasibility was determined by the successful completion of the grafting portion of the protocol. Compliance feasibility was determined by the participant’s ability to successfully complete the study from consent through the 12-month midpoint.
Postoperative evaluation
After DBS and PNT implantation, participants were admitted overnight and then followed clinically through routine postoperative visits, programming visits and for their postoperative study visits. Medication was reported as total daily levodopa equivalent dose (LED).24 For UPDRS testing preoperatively, while in the practically defined off-state, participants stopped antiparkinsonian medications at least 12 hours before undergoing UPDRS testing (>24 hours for long-acting medications). After surgery, participants, if on PD medication, underwent a minimum 12-hour withdrawal from antiparkinsonian medications and turned off the DBS stimulator for 12 hours before testing.
Clinical measures
Clinical measures included UPDRS (all components) preoperatively and at 6-month intervals postoperatively assessed by experienced raters blinded to location. Additional clinical measures included a comprehensive neurocognitive evaluation, Parkinson's Disease Questionnaire (PDQ) 8 measures, the Non-Motor Symptoms Scale and formal gait analysis. These measures were collected at baseline and at 24 months and will be presented at the completion of the study.
Data analysis
Study data were collected and managed using REDCap electronic data capture tools hosted at the University of Kentucky. Missing study visits (0 missing/18 visits at baseline, 0/18 at 6 months, 3/18 at 12 months) were not included as part of the analysis. Missing item data: scale items with missing data were imputed using mean substitution for that item across all participants at the corresponding time point. UPDRS Part IV questions 35–38 and 40–42 required a ‘yes’ or ‘no’ answer, which was scored with a 1 for yes or 0 for no. To address the missing data points for yes/no questions, we used the single imputation method. We randomly selected a participant, with a random number generator, who had a response and used that selected participant’s response as the missing measurement. Results are reported as mean (SD) unless reported as 95% CI.
Descriptive statistics were calculated for clinical outcomes. Linear mixed models were used to investigate changes in UPDRS III scores across time. Mixed models are appropriate where the independent observation assumption is violated, in this case through the inclusion of repeated measures, and are able to make use of all available data through the use of maximum likelihood estimation. An α level of 0.05 was used for significance of the omnibus test of time. Follow-up pairwise comparisons were controlled for multiple comparisons using the Tukey method. Data analysis was conducted using PROC MIXED as part of SAS V.9.4 (SAS Institute) and JMP Pro V.14.0 (SAS Institute).
Results
The data from 18 participants (table 1) are presented in this report and include the components of feasibility, safety, imaging, implant location and clinical outcome measures.
Participant demographics
Feasibility
We evaluated several aspects of study feasibility ranging from trial design to likelihood of successful surgery procedural completion. An overall summary is presented in figure 1. Twenty-one participants were enrolled; three of these were withdrawn on subsequent preoperative study visits: two because of the identification of additional, exclusionary clinical information and one because of family and social issues. All three went on to have successful DBS without PNT implants. All remaining participants (n=18) underwent the staged20 23 DBS-Plus protocol (mean time between stages: 7 days, range 3 days to 14 days). All participants were successfully implanted with a DBS system and subsequently received therapeutic programming and treatment. With respect to the PNT implantation protocol, all participants successfully received an implant without any procedural complications. In each case, the sural nerve was identified and transected during stage I, and the distal segment was identified and harvested during stage II. The amount of harvested tissue was adequate to provide enough tissue for fascicle dissection and implantation with enough left over for collection and storage in our tissue biobank.
The duration of the DBS-Plus surgical procedure, from incision to closure, averaged 208 min (30 min). All participants completed the 6-month evaluation and 15/18 completed the 12-month evaluation. One participant died, 7 months after surgery, from medical complications from a bowel obstruction. Two participants missed their evaluations for non-study related medical reasons (a hip fracture; a device-related infection).
Safety
The major objectives of this study were to assess the safety of the DBS-Plus surgical approach as well as the long-term safety and tolerability of the implanted PNT. With respect to the DBS-Plus procedure, there were no intraoperative complications from either the DBS portion of the surgery, or from the sural nerve transection during stage I, or from the nerve harvesting and delivery during stage II. The stage II procedure was well tolerated: 16/18 (89%) participants were discharged on post-op day one, one on day two and one on day three. Comparatively during the same time frame at our centre, for 33 consecutive patients with PD, 27/33 (82%) had a length of stay (LOS) of 1 day. Mean LOS for DBS-Plus=1.2 days; LOS for DBS only=1.6 days.
AEs (table 2) were adjudicated according to relatedness to the PNT intervention component of the protocol, which included the implant procedure and the implant itself. There were no serious AEs related to the study component. The only reported AEs related to the study intervention were an infection of the ankle incision following the second procedure and hypoaesthesia and hyperaesthesias of the lateral foot distal to the sural nerve tissue harvest site. The infection was superficial and was treated successfully with oral antibiotics. Altered sensitivity of the foot was reported in 6 of 18 participants and was an expected event given the nature of the procedure.
Adverse events rated as possibly, probably or definitely related to the study
Imaging
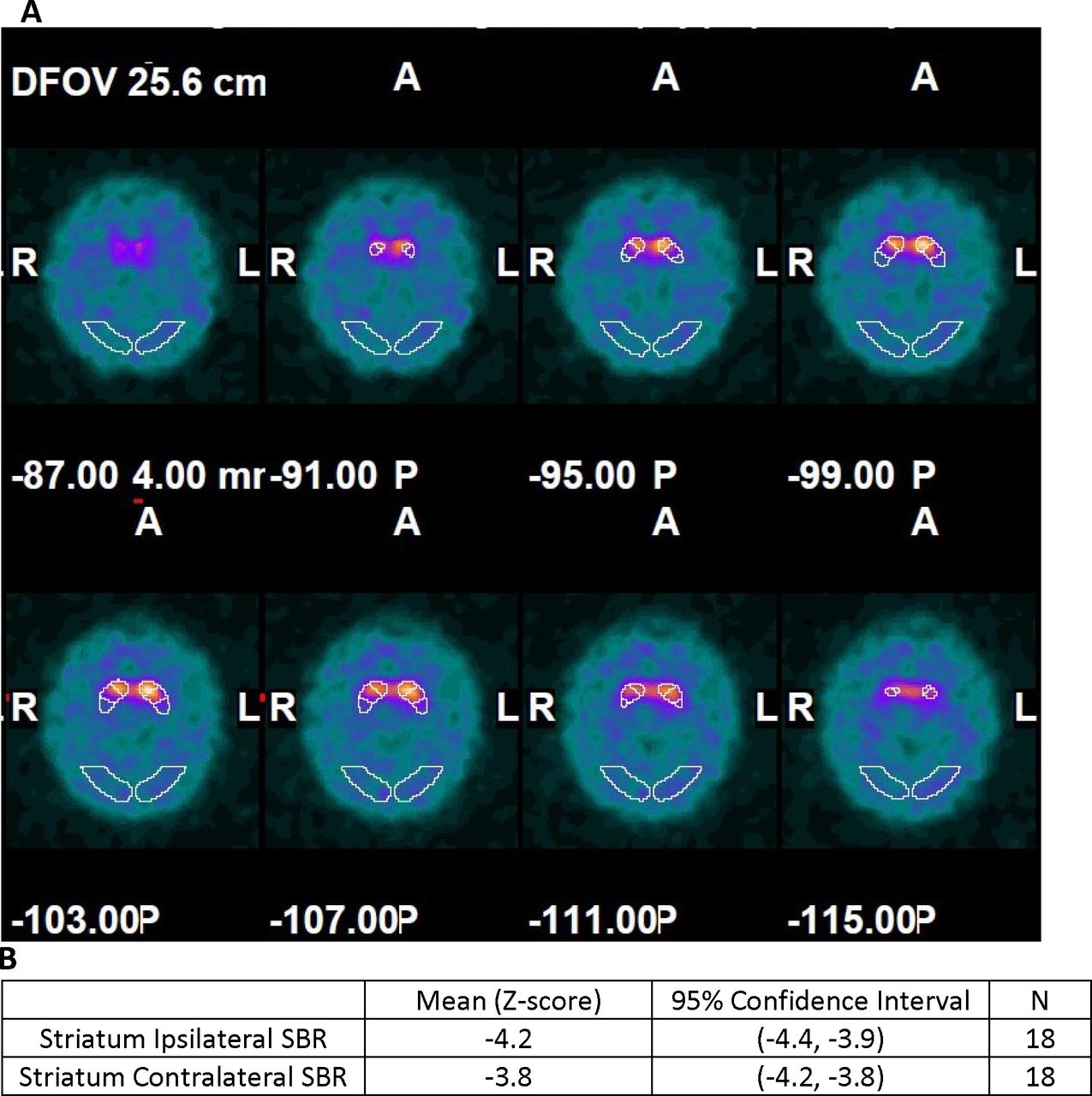
123I-ioflupane/SPECT imaging: all participants had scan results demonstrating loss of striatal signal intensity consistent with a presynaptic decrease in ligand binding (figure 3A) below levels of healthy control subjects (figure 3B). Thus, no subjects were excluded from PD diagnosis based on normal 123I-ioflupane/SPECT findings.
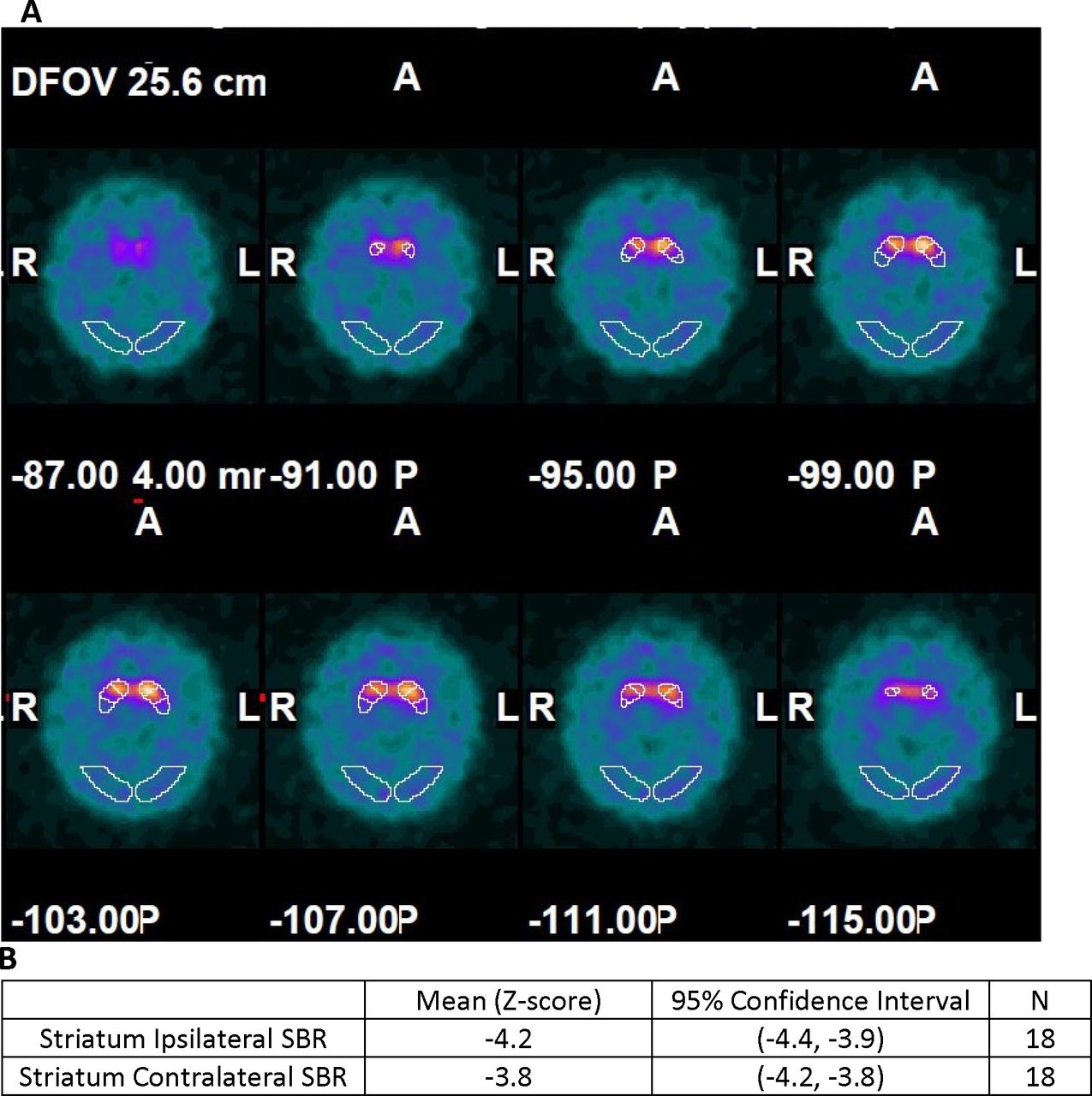
(A) Multiple depths through the axial plane show abnormal 123I-ioflupane binding in the outlined striatal regions in one study participant. Values indicate different depths oriented based on A (anterior), P (posterior), L (left) and R (right). (B) Baseline comparison of the mean specific binding ratio (SBR) (ipsilateral and contralateral to the intended peripheral nerve tissue implant location) to the DaTQUANT normal database.
Post-op MRI: clinical neuroradiological evaluations were performed on all 18 participants and reported typical postoperative changes associated with the DBS surgery—mostly mild pneumocephalus in several participants. One participant was observed to have a 1cc haemorrhage surrounding the DBS electrode but did not complain of any clinical symptoms or display any deficits associated with the finding; the participant was discharged on post-op day 1.
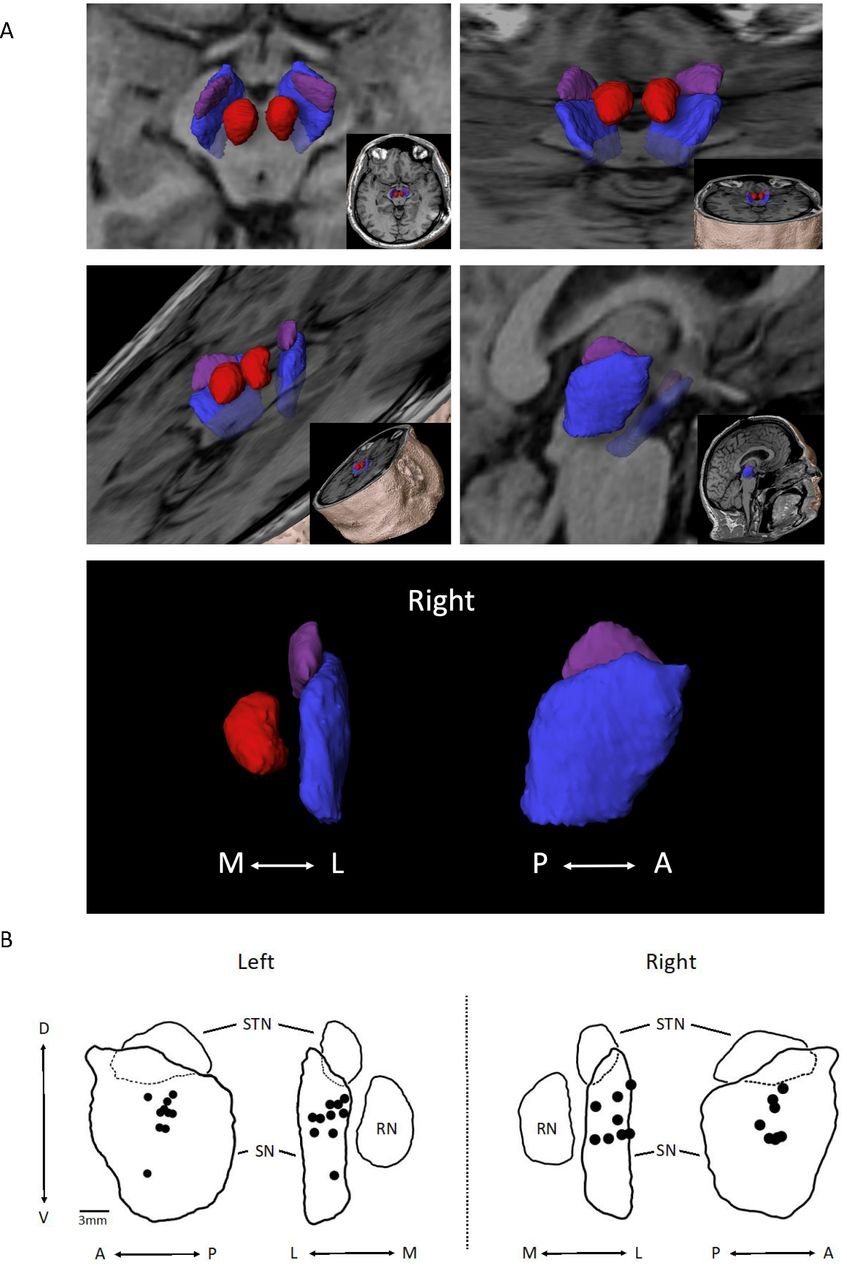
Assessment of PNT location
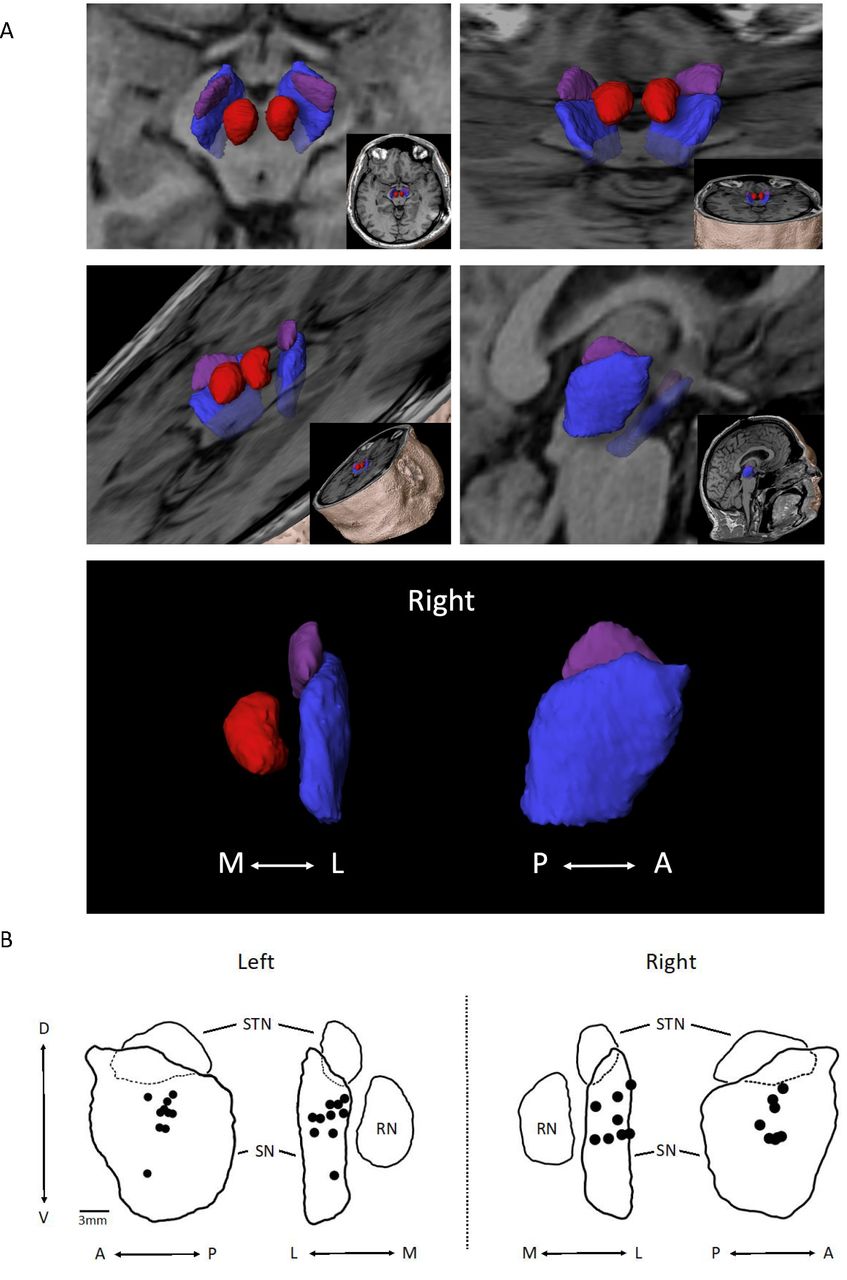
Implant locations were identified on the post-op MRIs and then mapped into the midbrain anatomical space in relation to the SN, red nucleus and STN (figure 4A). A composite of the centre point of the implant zones for each participant (figure 4B) shows that all implant zones were either fully within the SN (n=16) or on the border (n=2; lateral border of left SN). Within the SN, the grouping of implants was centred within the middle third (A–P), in the mid-half to upper-half (D–V)—except for one in the lower half, and distributed evenly (M–L).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) 3D object depiction of the target zone. Using MPRAGE images, space-filling objects of the subthalamic nucleus (STN) (purple), substantia nigra (SN) (blue), red nucleus (RN) (red) depicted in different planes are shown. (B) Peripheral nerve tissue (PNT) placement within the SN on the left (n=10) and right (n=8) are shown. Black dots represent the centre point of deployment as determined from analysis of post-op MRI sequences. Composites were organised from the analysis of individual PNT placements using Elements software. Outlines are of objects shown in (A). A, anterior; D, dorsal; L, lateral; M, medial; P, posterior; V, ventral.
Clinical measures
Table 3 shows secondary outcome analyses. Mean total LED was 899 (531) mg/day at baseline, 362 (301) mg/day 6 months after surgery, and 379 (259) mg/day 12 months after surgery. Following surgery, 2 of 18 participants reported continuing dyskinesias from presurgery although both participants showed improvement.
Participant outcomes
For UPDRS Part III practically defined off-state, scores decreased at 6 months after surgery and continued to be stably lower at 12 months after surgery. The overall main effect of time was significant F(2,17) = 9.73, p=0.002. There were significant decreases between baseline and 6 months (difference=−8.0, 95% CI −2.6 to −13.3 points, t=3.81, padj=0.004) and between baseline and 12 months (difference=−8.1, 95% CI −2.4 to −13.9 points, t=3.64, padj=0.005). The difference from 6 to 12 months was not significant (difference: 0.2, 95% CI −6.1 to +5.7 points, t=0.08, padj=0.996). To further explore the effects of unilateral implantation on the laterality subscores of the UPDRS, we compared lateral UPDRS items (items 20–26) at baseline and 12 months for the body sides contralateral and ipsilateral to the PNT graft. Mean (SD) lateral scores at baseline were 16.2 (5.4) points (contralateral side) and 10.1 (5.0) points (ipsilateral side). At 12 months, mean (SD) scores were 11.2 (3.9) points (contralateral side) and 9.5 (5.0) points (ipsilateral side). Mean differences at 12 months compared with baseline were −5.0 (95% CI −8.0 to −2.0) points for the contralateral side and −1.2 (−3.1 to +0.7) points for the ipsilateral side.
Discussion
In this report, we present a 12-month interim assessment of the feasibility, safety, with respect to subject participation and follow-through, and include available clinical outcome measures.
Feasibility
The requirements for participation were not burdensome as demonstrated by the high rate of completion of the evaluation visits. The PNT implant procedure did not interfere with the DBS surgery or therapy. All participants who were scheduled for surgery were able to receive bilateral DBS implant, followed by PNT placement, and all were successfully programmed in the clinic postoperatively.
Safety
The safety profile of the DBS-Plus procedure was favourable with respect to the surgical requirement of tissue harvesting and direct delivery to the SN. The postoperative LOS was no different than for DBS only patients implanted during the same time interval as the study (data not shown). No new neurological deficits occurred from the DBS surgery or the grafting procedure. Non-study related AEs were similar to those seen commonly with DBS surgery or were unrelated altogether.
From a surgical perspective, the sural nerve transection during stage I and harvesting during stage II are practically the same as performing a sural nerve biopsy twice, with the second biopsy occurring through the same incision 2 weeks following the first biopsy. There was no evidence of infection or poor wound healing related to this procedure.
Imaging
Radiological evaluations revealed expected postoperative changes typically related to DBS surgery. Postoperative images demonstrated accurate targeting of the electrodes. Based on previous intranigral deliveries,25–27 we targeted the midpoint of the rostral-caudal span of the SN. As shown in the composite of deposit locations, we successfully delivered to the SN. The dimensions of the PNT deposit were a cylinder approximately 5 mm in height and 1.6 mm in diameter. Depositing PNT at the midpoint of the medial-lateral axis (SN width ~4 mm) provided an opportunity for elements from the PNT to reach unhealthy dopamine neurons either within the interstitium or within the nigrosomes of the SN pars compacta.28 29
Clinical outcome
Clinical measures are important tools to assess the potential for worsening due to progression or an adverse response to PNT, or for possible improvements potentially related to the graft, grafting procedure (insertional effect) or placebo effect. As a phase I trial, this study is not designed or powered to determine clinical efficacy, but rather to evaluate clinical parameters and their ability to provide evidence for the possibility of disease modification. Overall, the clinical measures did not show any direct evidence of worsening or disease progression and performance scores remained generally stable. Although the complication of runaway dyskinesias has been associated with grafting of fetal dopaminergic tissue,30 we did not observe worsening of dyskinesias or development of new dyskinesias in our participants. Analysis of the UPDRS Part III off-state scores revealed a significant reduction in scores at 6 and 12 months compared with baseline. This finding not only provides strong evidence that the PNT implants did not hasten the disease progression, but also suggests that the procedure may hold some promise in the reduction of this outcome measure. Although a 12-hour washout period for medication and stimulation for the off testing may not be fully adequate to allow participants to reach a fully therapy-free state, keeping a standardised 12-hour protocol provides consistency and is not overly burdensome. Use of the UPDRS Part III as an endpoint in future studies will also require a strict definition of what constitutes a clinically important difference.
Evaluation of the clinical outcome measures demonstrates both strengths and weaknesses. The main utility of the majority of the clinical measures is to monitor for safety by having the ability to detect the possibility of acute worsening of disease symptoms. Considering we are surgically targeting the SN, the UPDRS III scores in the off-state remain the best tool for the assessment of changes in the motor components of the disease. Non-motor symptoms and manifestations most likely result from degeneration and cell loss from other nuclear regions.31–34
What are the next steps for PNT implantation?
Based on the outcomes reported here, we support a plan to move this project toward testing in a randomised, double-blind trial to assess the efficacy of PNT deployment to the SN. Furthermore, we think the DBS-Plus platform is a useful strategy for studying PNT implantation at this stage of development. However, despite the encouraging results reported here, fundamental issues remain to be resolved. Specifically, would additional PNT deliveries to the SN affect outcomes while remaining feasible and safe? As part of our methodical design, with safety and feasibility at the forefront, our next step is a ‘dose-finding’ study—multideployment unilaterally or bilaterally of PNT. The results from these types of studies would guide us in determining whether or not single-location, unilateral implantations, as described here, would be the dosage of PNT most appropriate to use in future efficacy trials. We chose to start with single delivery into the unilateral SN with the expectation that this approach would be the most feasible to perform with the least risk compared with multipass or bilateral deliveries. Therefore, our next steps will be to assess multilocation delivery of PNT to the SN unilaterally and bilaterally.
Summary
The results of this paper provide supporting evidence for the feasibility and safety of direct delivery of PNT into the SN of participants with PD at the time of DBS surgery. In our experience, the DBS-Plus approach offers many advantages for investigating novel interventional therapies involving patients with PD with advanced disease. We aim to provide transparency to allow this procedure or its components to be openly available to any interested teams. If successful, this type of approach could be used clinically in conjunction with standard DBS protocols. It could also be investigated for earlier implementation with the possibility of rescuing more of the ‘at risk’ cell population.35
Supplemental material
Data availability statement
No data are available.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by University of Kentucky IRB #44749. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank Dr. Richard Kryscio for his advice on imputations. We thank Morgan Yazell, Stephanie Morris and Renee Wagner for assisting with trial execution; Jaimie Hixson with data processing; and Mike Hilvers, Josh Patton, and Dave Chapman for assistance with the procedures.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Author contributions are listed below based on these three elements:1) Research project: A. Conception, B. Organization, C. Execution; 2) Analysis: A. Design, B. Execution, C. Review and Critique; 3) Manuscript: A. Writing of the first draft, B. Review and Critique. JEQ: 1B, 1C, 2A, 2B, 3A; JTS: 1A, 1B, 2C, 3B; JAG: 1B, 1C, 3B; CJM:2B, 3B; REK: 1C, 2C, 3B; MJC: 1C, 2C, 3B; ZG: 2C, 3B; GAG: 1A, 1B, 3B; CvH: 1A, 1B, 1C, 2A, 2B, 3A and is the guarantor for the overall content.
Funding This work was supported by gifts to the Brain Restoration Centre; Ann Hanley Parkinson’s Research Fund; Pro’s Players Fore Parkinson’s, the UK College of Medicine BRAIN Alliance, and the National Centre for Advancing Translational Sciences, through NIH grant UL1TR000117 and UL1TR001998.
Competing interests CvH has served as consultant to Boston Scientific and Brainlab.
Provenance and peer review Not commissioned; internally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.