Article Text

Abstract
Background Adherence and persistence are critical to optimising therapeutic benefit from disease-modifying therapies (DMTs) in relapsing-remitting multiple sclerosis (RRMS). This prospective, open-label, multicentre, observational study (AubPRO), conducted in 13 hospital-based neurology clinics around Australia, describes treatment satisfaction in patients newly initiated on teriflunomide (Aubagio) and evaluates the use of an electronic patient-reported outcome (PRO) tool.
Methods Patients (≥18 years) newly initiated on teriflunomide (14 mg/day) were followed up at 24 and 48 weeks. Patients completed questionnaires and pill counts electronically using MObile Data in Multiple Sclerosis. The primary endpoint was treatment satisfaction, measured by the Treatment Satisfaction Questionnaire for Medication (TSQM, V.1.4), at week 48. Secondary endpoints included treatment satisfaction at week 24, other PRO scales, clinical outcomes, medication adherence and safety.
Results Patients (n=103; 54 (52.4%) treatment naive) were mostly female (n=82 (79.6%)), aged 49.5 (11.8) years, with MS duration since symptom onset of 9.1 (11.8) years and a median Expanded Disability Status Scale score of 1.0. Mean treatment satisfaction scores were high (≥60%) across all domains of the TSQM V.1.4 at week 24 and at week 48. Compared with week 24, week 48 treatment satisfaction increased for patients who were treatment naïve and for those previously on another oral or injectable DMT. Over 48 weeks, PROs remained stable across a range of measures including disability, physical health, emotional health and mobility, and there were improvements in work capacity and daily life activity. Adherence was high throughout the study with mean compliance (pill counts) of 93.2%±6.26%, and 98 of 103 (95.1%) patients remained relapse-free.
Conclusion This cohort of Australian patients with RRMS, newly initiated on teriflunomide, and treated in a real-world clinical practice setting, reported high treatment satisfaction and adherence at 24 and 48 weeks. Patient-reported measures of disability remained stably low, work capacity and daily life activity improved, and most patients remained relapse-free.
- MULTIPLE SCLEROSIS
- MEDICINE
- ORAL MEDICINE
- QUALITY OF LIFE
Data availability statement
Data are available upon reasonable request. Research data are stored in an institutional repository and will be shared upon reasonable request and with permission of Sanofi Australia by emailing the corresponding author.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
In adult patients with relapsing-remitting multiple sclerosis, adherence and persistence are critical to optimising therapeutic benefit from disease-modifying therapies.
Electronic patient-reported outcome tools capture patient satisfaction and other treatment outcomes with teriflunomide in the clinical trial setting, but it is not known how this translates into the real-world setting.
WHAT THIS STUDY ADDS
Patients new to teriflunomide reported high levels of satisfaction/adherence.
This benefit extended to many areas of a patient’s life (disabilities, physical health, emotional health, mobility, work capacity/daily life activity) and corresponded with clinical observations.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE AND/OR POLICY
Electronically collected data reinforce the risk–benefit profile of teriflunomide and support the translation of clinical trial results into routine clinical practice.
Introduction
The majority of disease-modifying therapies (DMTs) used in patients with relapsing-remitting multiple sclerosis (RRMS) are viewed as long-term therapeutic options, necessitating that choices be highly individualised, accounting for disease stage, clinical activity, patient preferences and dosing/delivery practicalities.1 With up to 50% of people discontinuing therapy within 2 years,2 adherence and persistence are critical to attaining and optimising therapeutic benefit. Administration route,3 treatment satisfaction,4 side effects and convenience of treatment5 have been shown to be drivers of elevated adherence in patients with RRMS.
Using the validated Treatment Satisfaction Questionnaire for Medication (TSQM, V.1.46), treatment satisfaction with teriflunomide, a once-daily oral immunomodulator, has been demonstrated to be higher than that with interferon beta-1a in the clinical trial setting.7–9 The consistent efficacy of teriflunomide has been demonstrated in placebo-controlled clinical studies of patients with relapsing forms of MS10–12 and in those who experienced a first clinical episode suggestive of MS.13 Published data, evaluating teriflunomide under routine clinical practice conditions, report high levels of treatment satisfaction,14 15 with significant improvements observed in patients who had switched to teriflunomide from other DMTs.16
To our knowledge, no treatment satisfaction data are available for teriflunomide use in the Australian clinical practice setting where all registered DMTs are available as first-line treatment for RRMS. The study used a novel digital tool (MObile Data in Multiple Sclerosis (MOD-MS); RxMx, Sydney, Australia) to capture patient-reported outcome data and describe patient-reported satisfaction after commencing teriflunomide treatment in an Australian real-world setting.
Patients and methods
Study design and participants
This prospective, open-label, multicentre, observational study investigated real-world management of adult patients with RRMS who were newly initiated on teriflunomide. The study recruited patients attending hospital-based neurology clinics for routine outpatient consultations across 13 sites in Australia. The study was performed in accordance with the Declaration of Helsinki.17
Physicians identified patients eligible for treatment with teriflunomide, based on their own clinical practices, prior to and independently from study entry. After having decided to commence treatment, potentially eligible study participants were provided with study information. Eligible participants were: ≥18 years of age; had a documented diagnosis of RRMS; had not yet started treatment with teriflunomide; had no contraindications to and had not previously received teriflunomide; were not currently participating in an investigational interventional study; had access to a mobile web device with data/internet access or a personal computer (PC)/MAC browser with internet access; and were willing to complete the study questionnaires. Upon providing written informed consent, participants were enrolled into the study.
Enrolled patients attended clinic visits, scheduled according to patient-specific needs, and received treatment and evaluations for their MS as determined by their treating physicians, in accordance with local standards of care. Data were collected from patients and physicians (online supplemental figure 1). Patients completed self-reported questionnaires and pill counts electronically during the study, using validated MOD-MS software, a novel, digital application that enables automated platform-independent data collection with smartphones, tablets or PCs. Physicians collected clinical data using electronic case record forms during each of the three clinic visits, at baseline (visit 1), week 24 (visit 2) and week 48 (visit 3) after initiating teriflunomide.
Supplemental material
Assessments and measurements
At the baseline visit, demographic data, MS disease history, disability (objectively assessed using the Expanded Disability Status Scale (EDSS)), prior treatments and the existence of relevant concurrent conditions were documented. Patient-reported outcomes were assessed using five validated tools (table 1). These comprised: treatment satisfaction (TSQM V.1.46); disability worsening (Multiple Sclerosis Performance Scale (MSPS)18); impact on physical and psychological well-being (Multiple Sclerosis Impact Scale, version 2 (MSIS-29 V.2)19 20; Multiple Sclerosis Walking Scale, version 2 (MSWS-12 V.2)21 22); and impact on productivity (Health-Related Productivity Questionnaire, version 2 (HRPQ V.2)23). Physicians recorded clinical and safety observations at each study visit, including the number of relapses, concomitant medications and details of any adverse events. Treatment adherence was self-reported by patients, based on the number of missed tablets left in blister packs, and recorded electronically at 4-weekly intervals, beginning at the fourth week after the start of treatment.
Patient-reported outcome measures used in AubPRO
Study endpoints
The primary endpoint was treatment satisfaction, measured by TSQM V.1.4, with teriflunomide at weeks 24 and 48. Secondary endpoints were changes over time on other patient-reported outcome scales (MSPS, MSIS-29, MSWS-12 and HRPQ), the number of relapses during the study, treatment adherence over the study period and occurrence of adverse events.
Analyses
Patients were included in the trial based on their medical history and the decision to commence treatment with teriflunomide. It was planned to recruit 150 patients from 13 sites in Australia. The sample size calculation was based on the primary endpoint of TSQM V.1.4 and previously published estimated 95% CIs for the means of the four TSQM domains.24 25 The primary analysis population comprised all patients who satisfied the inclusion/exclusion criteria and completed the baseline visit, irrespective of whether they received teriflunomide. A safety population, defined as all patients in the primary analysis population who received at least one dose of teriflunomide, was used for safety endpoint analyses. Adverse events were graded for severity, using a 3-point scale (mild, moderate, severe), and classified based on causality, seriousness and special interest.
All analyses were descriptive in nature and were completed based on available data; no imputation algorithms were applied for missing data and the patient-reported outcome questionnaires used were scored per published instructions. All data for patients who withdrew early were used for the analysis, up to the point of their discontinuation. Spearman non-parametric correlation was used to quantify relationships between TSQM domains and other variables. Selected patient-reported outcomes (MSPS, MSWS-12 and MSIS-29) were analysed with repeated measurements analysis of variance to assess the effect of each domain over time. The repeated measurements model included the scores at baseline, week 24 and week 48. For all endpoints, where statistical testing was conducted, the hypothesis was that mean score would be significantly different (p<0.05) at one or more time points (namely baseline, week 24 and week 48). All statistical tests were two sided at the 5% significance level. All analyses were conducted using SAS V.9.4 (SAS Institute).
Results
Across the 13 study sites, 105 patients consented to participate. Demographic and baseline disease characteristics are summarised in table 2. Two enrolled patients were later found to not meet the inclusion criteria and were excluded from the primary analysis population, a further three patients did not receive teriflunomide and were excluded from the safety analysis population.
Subject disposition, demographics and baseline disease characteristics
Over two-thirds of patients (71 of 103; 68.9%) underwent baseline MRI as part of their standard of care monitoring. Gadolinium-based contrast was used in 43 (60.5%) patients, of whom 17 were found to have gadolinium-enhancing lesions. A total of 54 (52.4%) patients were treatment naïve. Of the 49 patients who had previously been treated, the most recent DMTs, taken before study entry, were glatiramer acetate (15, 30.6%), beta-interferons (13, 26.5%), fingolimod (11, 22.4%), fumaric acid (8, 16.3%), daclizumab (1, 2.1%) and natalizumab (1, 2.1%). Prior to their most recent therapy, 20 patients had received multiple other DMTs, including beta-interferons (n=18), glatiramer acetate (n=9), fingolimod (n=3), natalizumab (n=3) and fumaric acid (n=2).
Primary outcome: treatment satisfaction
Mean (SD) treatment satisfaction scores were high across all four domains of the TSQM at both weeks 24 and 48. The highest satisfaction scores were for convenience (week 24: 87.48 (14.90); week 48: 88.97 (12.25)) and side effects (week 24: 79.52 (25.96); week 48: 83.61 (22.44)), with similar results observed for effectiveness (week 24: 63.19 (18.09); week 48: 63.85 (19.50)) and global satisfaction (week 24: 59.55 (25.00); week 48: 64.51 (23.86)). A scatter plot of individual TSQM scores in each domain revealed close similarities between week 24 and week 48 within each domain (figure 1). The TSQM scores in the convenience domain were all above 50 with the exception of one outlier at week 24. The scores in the other domains were more widely distributed, with a trend for higher (better) satisfaction scores and fewer outliers in the side effects and global satisfaction domains at week 48 vs week 24.

TSQM treatment satisfaction scores at week 24 and week 48: individual scatter plots by TSQM domain. TSQM, Treatment Satisfaction Questionnaire for Medication.
Compared with week 24, at week 48, mean TSQM scores were improved in the effectiveness, convenience and global satisfaction domains in patients who were treatment naïve and in the side effects and global satisfaction domains for patients who were previously on either another oral medication or an injectable DMT (online supplemental table 1). There were no significant correlations between the number of relapses in the last year and any of the four TSQM domains (online supplemental figure 2A). The number of missed doses between weeks 4 and 24 was negatively correlated with global satisfaction at week 24 (r-squared =−0.4098, 95% CI −0.5790 to –0.2067, p=0.0001) and with side effects at week 24 (r-squared=−0.3568, 95% CI −0.5855 to –0.0755, p=0.0118) (online supplemental figure 2B).
Secondary patient-reported outcomes
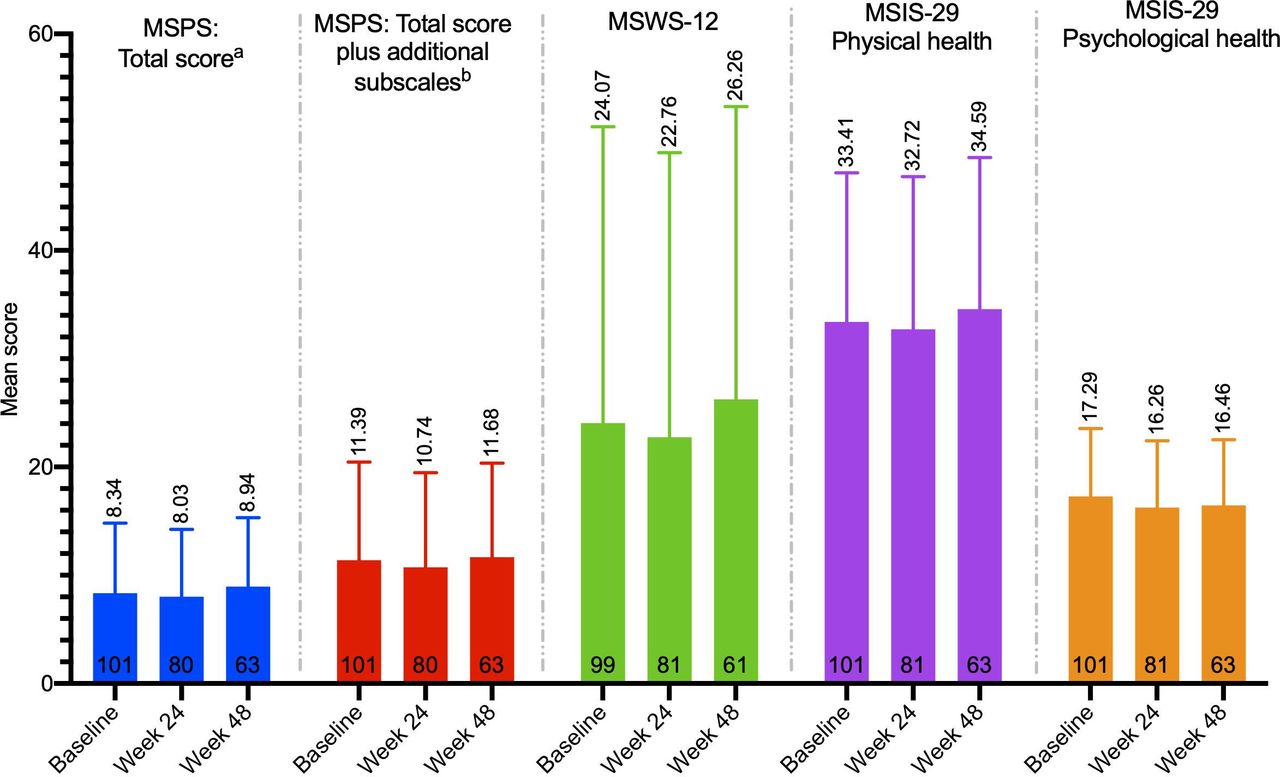
Patient-reported disability was low and remained stable from baseline to week 48, as measured by the MSPS total score and the MSWS-12 (figure 2). Physical and psychological health, as measured by the MSIS, also remained stable throughout the study (figure 2). Within the individual subscales of the MSPS (online supplemental figure 3), the majority of patients reported normal/minimal or mild/moderate disability at both baseline and week 48 and the proportion of patients reporting severe/total disability decreased from baseline to week 48 in four of the eight MSPS subscales (mobility, vision, fatigue, sensory symptoms) and in all three of the additional subscales (pain, depression, tremor/coordination).
{kind=link}
{kind=link}
Mean (SD) patient-reported outcome scores over time.* *A repeated measures ANOVA was used to assess the effect over time; all p values were non-significant (p>0.05). Higher scores indicate worse disability; maximum scores: MSPS total score=41, MSPS total score with additional subscales=66, MSIS-29=100, MSWS-12=100. aSubscales: mobility, bowel/bladder, fatigue, sensory, vision, cognition, spasticity and hand function. bSubscales: mobility, bowel/bladder, fatigue, sensory, vision, cognition, spasticity, hand function, pain, depression and tremor/coordination. ANOVA, analysis of variance; MSIS-29, Multiple Sclerosis Impact Scale; MSPS, Multiple Sclerosis Performance Scale; MSWS-12, Multiple Sclerosis Walking Scale.
Work capacity and daily life activity, as measured by the HRPQ, revealed improvements in multiple areas at week 48 compared with baseline (online supplemental table 2). The mean number of hours of work lost due to absenteeism reduced (5.63±13.61 hours at baseline vs 1.08±5.02 hours in the week before the week 48 visit) and there was an increase in the mean number of hours worked (25.92±16.48 hours at baseline vs 28.51±12.80 hours in the week before the week 48 visit), with a corresponding overall reduction in the percentage of work lost due to presenteeism and absenteeism combined (11.64±27.30 at baseline vs 6.71±16.35 in the week before the week 48 visit). The most notable improvement was in full-time employees, who reported a 70% reduction in work loss at week 48 vs baseline (percentage of work lost due to presenteeism and absenteeism combined: 21.94%±36.85 at baseline vs 6.69%±18.23 in the week before the week 48 visit). Similar trends were observed for household hours worked (online supplemental figure 4).
Clinical and safety observations
The majority of patients (98 of 103; 95.1%) remained relapse-free during the study period; half of these patients (55 of 98; 56.1%) had evidence of active disease (defined as one or more relapses in the year before the study and/or the presence of gadolinium-enhancing lesions on MRI prior to the baseline visit) prior to starting treatment. Five (4.9%) patients each experienced a single clinical relapse during the study period; four were treatment naïve and one had previously been treated with interferon beta-1b. These patients were younger (36.8 vs 50.1 years) and more likely to have had a relapse in the year prior (4 of 5 (80.0%) vs 55 of 98 (56.1%)) than those who did not relapse; relapse rate was higher in men than in women (3 of 21 (14.3%) vs 2 of 82 (2.4%)).
Of the 100 patients who received treatment, 69 (69.0%) completed the study as planned. Reasons for study withdrawal included adverse events (n=17, 17%), other reason (n=4, 4%), lack of efficacy (3, 3%), investigator decision (n=3, 3%), lost to follow-up (n=2, 2%), disease progression (n=1, 1%) and intolerance (n=1, 1%). Adherence with teriflunomide dosing was high throughout the study. The majority (80 of 97, 82.5%) of patients reported >95% compliance and the mean overall compliance (actual number of doses divided by expected number of doses×100) was 93.2%±16.26%.
Three-quarters of patients (75 of 100, 75.0%) reported at least one adverse event, and in over half (58 of 100, 58.0%) these were treatment related (table 3). Nine serious adverse events were reported during the study period by six patients (6.0%); none were considered treatment related and none were adverse events of special interest. Twenty patients (20.0%) discontinued treatment due to an adverse event; 10 (50%) of the withdrawals were at one site, 2 each at three sites and 1 each at four sites. Events occurring in more than one patient were diarrhoea, alopecia, nausea and rash (table 3). Four patients (4.0%) experienced an adverse event of special interest: increased alanine aminotransferase (n=2), hypoaesthesia (n=1), and one hypertension (n=1). No deaths were reported during the study.
Summary of adverse events (AEs) occurring in more than 4% of patients
Discussion
This observational, prospective study documents treatment satisfaction in adult patients with RRMS newly initiated on teriflunomide under routine clinical practice conditions. It adds to the growing body of patient-reported outcomes data in the real world,14–16 providing insights from the Australian setting and demonstrating the utility of the novel MOD-MS software for digital data collection.
The study population comprised mostly older (49.5 years) patients with long disease duration (9.1 years), which is more consistent with other teriflunomide real-world trials (TAURUS-MS14 and Teri-PRO15) than is the case with the teriflunomide TEMSO10 and TOWER12 clinical registration trials. The study population had a lower median baseline EDSS score (1.0) and a higher proportion of treatment-naïve patients (52.4%), compared with other teriflunomide real-world trials.
Patients reported high levels of treatment satisfaction, after commencing treatment with teriflunomide, across all four domains of the TSQM. The mean week 48 TSQM score data in this study are comparable with those previously reported in the global phase 4 Teri-PRO Study15 for effectiveness (63.85 vs 66.3), side effects (83.61 vs 84.1), convenience (88.97 vs 90.4) and global satisfaction (64.51 vs 68.20). Minimum clinically important mean values for the TSQM domains have not been published but the results show that the scale midpoint has been exceeded in all four domains, indicative of high levels of treatment satisfaction, sustained throughout the 48-week study period.
The performance validity and reliability of the TSQM have been established in patients with RRMS.7 Consistent with this observation, strong correlations were found among TSQM domains (online supplemental figure 1A), supporting the robustness of the data, despite the relatively small number of patients included. There were significant, negative correlations between the number of missed doses, during weeks 4–24, and the TSQM side effect and global satisfaction domains at week 24, these two variables decreased as the number of missed doses increased.
The literature supports associations between higher levels of treatment satisfaction, increased compliance and improved clinical outcomes.5 In accordance with this, clinical observations demonstrate high levels of adherence (>95% in the majority of patients) and treatment persistence (65.7% remained on teriflunomide treatment at the end of the study) and good clinical outcomes with >95% of patients remaining relapse-free, despite over half having evidence of active disease prior to starting teriflunomide. Teriflunomide was well tolerated and the safety profile of teriflunomide was consistent with that seen in phase 2, phase 3 and other phase 4 clinical studies. Twenty patients withdrew from treatment due to adverse events; diarrhoea (six patients), alopecia (four patients) and nausea (three patients) were the most frequent adverse events. Half of the patients who withdrew were from one site; this site accounted for half of the alopecia (two of four) and diarrhoea (three of six)-related withdrawals but was also responsible for 28% of patients recruited into the study.
This observational study was open label, not randomised and did not include a control group. The inclusion and exclusion criteria were established in line with the approved indication for teriflunomide in Australia. As an observational study, a bias, based on disease severity and label indication, may have existed which could influence patient selection by the physician and risked recruiting a non-representative study sample. Although the mean age and gender demographics of the study cohort are similar to those reported in other real-world studies in Australia,26 27 the baseline EDSS scores indicate a population with mild disease (85% with a baseline EDSS of 0–3). This is concordant with the treatment history which showed that 52% of the patients were treatment naïve and that the remainder had been switched to teriflunomide from a low efficacy agent.
Strategies for initiating DMTs are tailored to the individual patient but, in general terms, follow either a step-up strategy in response to disease progression or a step-down approach in response to disease stability.28 The initiation of teriflunomide in this cohort therefore represents a step-up approach. Key demographic characteristics of the patients in this cohort closely relate to those identified in a recent clinical consensus, which suggested suitability of teriflunomide in adult patients, including women on effective contraceptives, who were either treatment naïve or switching from lower efficacy treatments and who had mild-to-moderate disease activity.29 The results of the study are generalisable to an Australian population of patients with MS who met the prescribing criteria and were selected for teriflunomide therapy by their physician. While they may not be reflective of outcomes in the wider, general population with MS, the demographic profile of the cohort provides confidence that the results may be generalisable to a population of patients with MS in whom a step-up treatment strategy is being considered.
Consistent with the general population with MS,30 the ratio of women to men in our cohort was 4:1. The age range of the women in our cohort was wide (20–73 years), but more than half (56%, 46 of 82) were aged 50 years or more. Treatment choice in women is influenced by disease activity, reproductive age and family planning.31 Due to potential teratogenicity, reliable contraception must be used in conjunction with teriflunomide and, should pregnancy occur, an accelerated elimination protocol with colestyramine can be used.28
Despite limited generalisability to the wider population with treatable MS, this cohort of patients reported low levels of disability, which remained stable over the study period, and improved productive work life. These findings support patient-perceived benefits of teriflunomide that extend into many areas, including disabilities, physical health, emotional health, mobility and work capacity/daily life activity. The most notable reduction in absenteeism/presenteeism was in patients who were in full-time employment. This is in concert with prior data showing that effective management of MS symptoms contributes to patient productivity and enables them to continue working for longer.32 This is of importance, given recent Australian research which reported a reduction of 14.2% in productive work time and related $A6767 loss per person annually.33
Currently, there are 14 DMTs licensed in Australia for patients with RRMS: five injectable therapies (glatiramer acetate, interferon beta-1a, interferon beta-1b, peginterferon beta-1a, ofatumumab), six oral therapies (cladribine, dimethyl fumarate, fingolimod, ozanimod, siponimod, teriflunomide) and three administered via infusion (alemtuzumab, natalizumab, ocrelizumab). Within the Australian setting, access to this full range of DMTs is restricted only to the licensed indications, with no hierarchical or formally mandated lines of therapy. This wide choice does not reduce the burden of decision-making. The usefulness of patient-reported outcome tools, as a means of understanding the effects that MS and its treatment have on patients’ lives, has been expanded beyond the clinical trial setting.34 35 Such measures bring a unique perspective to what really matters to patients. Recognising that clinicians and patients may have differing opinions, the use of patient-reported outcome measures might be of value to help highlight patients’ unmet needs and focus clinical discussions.29 Despite this positive position, it is recognised that challenges remain in the uptake of these measures in decision-making processes.36 37 The ability to collect patient-reported outcomes electronically has been highlighted as a core consideration of the adoption of patient-reported outcomes into clinical practice.37 Recognition that the use of patient-reported outcome questionnaires in the clinic is limited by time constraints and staff workloads has led to the development of MOD-MS, a custom-designed browser-based cross-platform software tool, to collect patient-reported outcomes on a smartphone, tablet or PC. MOD-MS has been specifically designed to automate phase IV observational studies, using a customisable library of validated e-questionnaires that are automatically pushed to patient devices at predefined study time points, with real-time data capture, scoring/analysis and aggregated database population. MOD-MS delivers questionnaires through personalised time-sensitive web-links (sent by email and/or SMS) and facilitates their completion through customisable reminders that are configured at study start-up. No specific software is required by study participants. The results of the current study further demonstrate the feasibility of using patient-reported data capture in the real-world setting. Wider application of this tool may help to further enhance workflow efficiency in the MS clinic and improve patient outcomes by enabling early detection of changes in treatment satisfaction and medication adherence.
Conclusion
This cohort of Australian patients with RRMS, newly initiated on teriflunomide and treated in a real-world clinical practice setting, reported high levels of treatment satisfaction and adherence at 24 and 48 weeks. The majority of patients remained relapse-free over the 48-week therapy period, patient-reported disability outcomes remained low and stable, and work capacity and daily life activity were improved. These electronically collected observational data reinforce the risk–benefit profile of teriflunomide and demonstrate that findings from the clinical trial setting can be translated into routine clinical practice.
Data availability statement
Data are available upon reasonable request. Research data are stored in an institutional repository and will be shared upon reasonable request and with permission of Sanofi Australia by emailing the corresponding author.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and ethics approval was granted by three committees to cover the different study sites as follows: Austin Health Human Research Ethics Committee (HREC/16Austin/76), Calvary Health Care Adelaide Human Research Ethics Committee (16 CHREC E005) and UnitingCare Health Human Research Ethics Committee (2016.21.199). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors wish to acknowledge the patients who participated in this research, clinical study management support provided by Bonnie Emerson (Sanofi Australia) and patient support provided by Karen Thomas (Royal North Shore Hospital). Julie Hill, McCloud Consulting Group, conducted statistical analyses and Hazel Palmer of Scriptix, provided professional writing assistance in the preparation of this manuscript. Sanofi Australia funded both.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors TH—investigation, resources, writing (review and editing). JP—conceptualisation, methodology, investigation, resources, writing (original draft preparation). HB—conceptualisation, methodology, software, investigation, resources, writing (review and editing). SB—investigation, resources, writing (review and editing). RM—investigation, resources, writing (review and editing). RGB—investigation, resources, writing (review and editing). NS—investigation, resources, writing (review and editing). AL—investigation, resources, writing (review and editing). WC—investigation, resources, writing (review and editing). CS—investigation, resources, writing (review and editing). RW—conceptualisation, methodology, software, formal analysis, writing (review and editing). JM—validation, resources, project administration, writing (review and editing). MS—formal analysis, project administration, writing (review and editing). MB—conceptualisation, methodology, software, investigation, resources, writing (review and editing). SV—conceptualisation, methodology, investigation, resources, writing (original draft preparation), guarantor of the article. All authors approved the final version of the article.
Funding This study was funded by Sanofi Australia (study number: TERRIFL0776).
Disclaimer The authors were responsible for all content, interpretation of the data and the decision to publish the results as per the contributorship statement; they received no honoraria related to the development of this manuscript. The sponsor funded statistical and editorial assistance and reviewed the final draft before submission. Statistical analyses were performed externally (Julie Hill, McCloud Consulting Group). Editorial assistance in the preparation of this manuscript was provided by Hazel Palmer of Scriptix.
Competing interests TH has received speaking fees or received honoraria for serving on advisory boards for Biogen, Merck, Teva, Novartis, Roche, Bristol Myers Squibb and Sanofi. He is co-editor of Advances in Clinical Neurosciences and Rehabilitation. JP reports grants from Roche, Merck, Sanofi, Biogen and consulting agreements for Roche, Merck, Sanofi, Biogen and Eli-Lilly. MB reports grants from Biogen, grants from Novartis Pharmaceuticals, grants from Sanofi, grants from Merck, outside the submitted work; and MB is a consultant to RxMx and research director at the Sydney Neuroimaging Analysis Centre. HB reports personal fees and other from Biogen, personal fees and other from Novartis, personal fees and other from Sanofi, personal fees and other from Roche, personal fees and other from Merck, outside the submitted work. AL, RGB, RM, NS, WC and CS have nothing to disclose. SB reports grants and speaker fees from Merck, Biogen, Sanofi, Novartis, Bayer, Otuskua, CSL and Roche. MS is an employee of Sanofi and may hold shares and/or stock options in the company. RW is an employee of Sanofi and may hold shares and/or stock options in the company. JM is an employee of Sanofi and may hold shares and/or stock options in the company. SV reports personal fees and other from Biogen Idec and Merck, outside the submitted work.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.