Article Text

Abstract
Objectives To assess the effectiveness of Neuroaspis plp10 nutritional supplement when added to interferon (IFN)-β treatment in patients with relapsing-remitting multiple sclerosis (RRMS).
Design A 30-month phase III multicentre, randomised, double-blind, placebo-controlled trial. Randomisation stratified by centre using a computer-generated procedure with Neuroaspis plp10 versus placebo in 1:1 ratio. The first 6 months were used as both the pre-entry and normalisation period.
Setting 3 teaching hospitals in Greece and 1 Neurology Institute in Cyprus.
Participants 61 patients with RRMS on IFN-β were randomly assigned to receive Neuroaspis plp10 (n=32) or placebo (n=29), 20 mL, orally, once daily, for 30 months.
Intervention Neuroaspis plp10, a cocktail mixture, containing specific PUFA (12 150 mg) and γ-tocopherol (760 mg) versus virgin olive oil (placebo).
Main outcome measure The primary end point was the annual relapse rate (ARR) whereas the secondary ones were the rate of sustained progression of disability, as measured by the Expanded Disability Status Scale (EDSS) and the brain T2 and gadolinium-enhancing lesions, at 2 years.
Results For the intention-to-treat analyses Neuroaspis plp10 significantly reduced the ARR by 80%, (RRR, 0.20; 95% CI: 0.09 to 0.45; p=0.0001) and the risk of sustained progression of disability by 73% (HR, 0.27; 95% CI: 0.09 to 0.83; p=0.022) versus placebo, at 2 years. The number of T1 gadolinium-enhancing lesions and the number of new/enlarged T2-hyperintense lesions were significantly reduced (p=0.01 and p<0.0001, respectively). Both T1-enhancing and new/enlarging T2-hyperintense lesions were significantly reduced (p=0.05 and p<0.0001, respectively). No significant adverse events were reported.
Conclusions Neuroaspis plp10 added to IFN-β was significantly more effective than IFN-β alone in patients with RRMS.
Trial registration number ISRCTN06166891.
- MULTIPLE SCLEROSIS
- MEDICINE
- PARKINSON'S DISEASE
- QUALITY OF LIFE
- RANDOMISED TRIALS
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Eicosapentaenoic acid/docosahexaenoic acid (omega-3) and linoleic acid/γ-linolenic acid (omega-6) specific structural essential oil molecules are known for many years for their properties and relation to the biomechanisms involved in autoimmune, inflammatory, neurodegenerative chronic diseases but without real proof of significant efficacy.
Most patients prefer using supplementary and or alternative medical products because the existing disease-modifying therapies are associated with side effects and limited efficacy.
WHAT THIS STUDY ADDS
This is the first well-studied formulation (Neuroaspis plp10) based on specific nutritional essential oil molecules and specific vitamins that have been tested by different clinical studies, now with a small randomised, double-blind phase III trial and especially through a robust well-defined protocol.
For the first time, a nutritional formulation has been proved to promote significantly reduced annual relapse rate and the risk of sustained disability progression, supported by MRI findings and without any serious side effects.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study might affect the overall practice and policy for the use of specific suppliments and nutraceuticals in the treatment and prevention of different serious and or chronic diseases when are appropreatly formulated and tested.
Introduction
Relapsing-remitting multiple sclerosis (RRMS) is characterised by the development of inflammatory lesions in the central nervous system (CNS), resulting in demyelination and axonal loss.
Many different dynamic processes are involved and simultaneously coordinated for MS pathogenesis. Oligodendrocytes, the myelin-forming cells of the CNS, are target cells in the pathogenesis of MS, but the exact aetiology is unknown. Pathological mechanisms that seem to be involved in MS include immune-mediated inflammation, oxidative stress and excitotoxicity.1 These mechanisms can potentially contribute to oligodendrocyte and neuronal damage and cell death, leading to disease progression.
Polyunsaturated fatty acids (PUFAs), such as the omega-3 eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), are the most abundant structural components of the neural tissue and play a fundamental role in the development and the proper functioning of the nervous system. The PUFA composition of membrane phospholipids are involved in a variety of cellular and multicellular processes, including inflammation and immunity, with implications to diseases such as MS. The fatty acid composition of phospholipids determines the biophysical and functional characteristics of the cellular membranes such as ‘fluidity’, transport, polarity and plays an important role in cellular integrity and intracellular and intercellular communication.2
PUFA and antioxidant deficiencies have been reported in patients with MS.3 Both antioxidants, vitamin E and γ-tocopherol are efficiently implicated in trapping reactive oxygen and nitrogen oxide radicals,4 respectively, and both exert non-antioxidant properties, including modulation of cell signalling, regulation of specific gene transcription, modulation of immune function and induction of apoptosis.5 6
In vitro, in vivo and ex vivo studies have demonstrated that diet EPA/DHA omega-3 and omega-6 linoleic acid (LA)/γ-linolenic acid (GLA) may be implicated and modulate many of the known complex pathways in MS pathophysiology. Anti-inflammatory properties of omega-3 and omega-6 PUFAs include competitive inhibition of arachidonic acid (AA) and its metabolites that can be involved in promoting inflammation7; they promote production of anti-inflammatory prostaglandins, thromboxanes and can inhibit production of proinflammatory cytokines8; they promote reduction of the level of the proinflammatory interleukin-1, interleukin-1-α and tumour necrosis factor.9 10 They have also been reported to produce lipoxins and modify the cytoskeletal components, thereby inhibiting the ability of leucocytes for migration.11 12 Omega-3 PUFAs inhibit the expression of the nuclear transcription factor, κ-B that is involved in the production of inflammatory cytokines, chemokines and various adhesion molecules with crucial roles in MS.13–15 Resolvins and protectins are derived from omega-3 PUFAs through lipooxygenase-mediated mechanisms16 17 and promote control of inflammation in neural tissues by activating G-protein-coupled receptors,18 inhibition of neutrophil, reduction of tumour necrosis factor expression, interferon (IFN)-α production and T-cell apoptosis.17 In vitro, T-cell proliferation in acute and chronic inflammation can be reduced by supplementation with either omega-6 or omega-3 PUFA.19 Furthermore, DHA prevented dendritic cell maturation and CD4+ T cell stimulation and differentiation, in an animal model of MS.20
Culture cell-line experimental studies report that cyclooxigenase-2 participates in the formation of electrophilic fatty acid derivatives of the omega-3 fatty acids in activated macrophages21 which in turn are implicated in the activation of the nuclear respiratory factor 2 that induces the transcription of a number of protective genes. Τhe electrophilic fatty acids activate the peroxisome proliferator-activated receptor (PPAR)γ for anti-inflammatory response. Omega-3 PUFAs and their eicosanoid derivatives are thought to possess strong PPAR-α-agonist properties on T cells in humans. This event has been shown to be neuroprotective in experimental animal models.22–24
Animal studies report that retinol X receptor (RXR)γ is a positive regulator of the endogenous oligodendrocyte precursor cell differentiation and remyelination.25 EPA and DHA have neuroprotective effects, are endogenous ligands of RXRs and PPAR and have been found to increase neurogenesis in old rats.26 27 DHA may block depolarisation-induced increased glutamate efflux and the activation of glutamate receptors leading to excitotoxicity, partly through inhibition of voltage-sensitive Na+ and Ca2+ channels.28 In vitro, omega-3 PUFAs have been shown to prevent neuronal accumulations of Ca2+, which can trigger a destructive cellular cascade of events that leads to neuronal damage and death.28 In vitro, omega-6 PUFAs can alter the function of oligodendrocytes by affecting their membrane composition.29 Membrane depolarisation affects protein phosphorylation of myelin basic protein in oligodendrocytes, an important event in myelination.30 DHA is reported as neuroprotective against excitotoxicity, inflammation and oxidative stress.28 Recent data report that dietary intake of a preformed DHA supplement is more effective in reaching the brain and achieving neuroprotection in an animal model of PD.31 PUFAs might also interfere with the production of certain matrix metalloproteinases that can be the cause of disruption of the brain–blood barrier,32 but they can also stimulate the production of molecules involved in myelinogenesis.33 Thus, the action mechanisms of the omega-3 PUFAs may most likely be considered as holistic, multifactorial and may be related to a number of cellular and molecular effects in CNS.
At present, agents used in MS seem to be partially effective and lack remyelinating and significant neuroprotective properties. Moreover, severe side effects have been reported to be associated with current RRMS treatments.34
The increasing incidence and prevalence of MS and the long-term sequelae of the disease urges the need for the development of new treatments to prevent activity and disability progression in patients with this condition. Based on the above research findings, the afore-mentioned specific dietary antioxidants and specific omega-3 and omega-6 PUFA fulfil the biological criteria and have the potential to affect disease activity and progression.1 35 36 The inconsistent data available from clinical trials performed to date on PUFA dietary interventions as complementary therapies for MS are the result of several notable limitations including supplement optimal dosing, design and selection of outcome measures.37 38
In 2013, we have published the results of a proof-of-concept clinical trial39 based on a dietary cocktail formula preparation using the exact same ingredients, formulation and dosing as now named ‘Neuroaspis plp10’ dietary intervention, with promising outcome and statistically significant; reducing the annual relapse rate (ARR) and the risk of sustained disability progression. With this study, we are reporting the results of a phase III, multicentre, double-blind, randomised, placebo-controlled clinical trial of efficacy and safety, the MINERAL Study, using the Neuroaspis plp10 dietary supplement.
Methods
Patients
According to the study protocol, for a two-sided type I error (α) of 0.05 and 80% power with a possible 35% dropout, 220 patients (110 per treatment arm) would be needed to be enrolled. That power calculation incorporates a Bonferroni adjustment in order to account for there being two primary end points. The sample size is based on the efficacy of IFNs on the MRI lesions and assumes that plp10 will reduce a 2-year percentage of patients with disease progression by 60%. Finally, it assumes that the time to disability progression will be analysed via the Cox proportional hazards model.
However, during the enrolment process, changes in the international clinical practice guidelines made it difficult to enrol patients in accordance with the study protocol. Thus, a post-hoc power calculation was performed in order to readjust the study sample size based on the current enrolment rate and using the ARR as the model input. Assuming an ARR for the intervention group equal to 0.4 and for the control group equal to 1.04, 27 patients per group would be required in order to achieve 80% power for a two-sided type I error (α) of 0.05 based on a Poisson model.39 40 Out of 61 eligible patients, 32 patients were randomly assigned to receive Neuroaspis plp10, a cocktail mixture of specific PUFA and γ-tocopherol and 29–205 receive placebo from January 2016 to 31 December 2018. The first 6 months for each enrolled patient was used as the normalisation period, while the rest 24 months was considered as the main study duration. All patients gave written informed consent.
The study protocol (ISRCTN06166891) was developed by the investigators. Study data per patient were reported and filed by the assigned examining/treating physicians at the involved sites through the central trial data-collection platform designed exclusively for the trial using a specific confidential per site-access-code. Data from all sites were finally received by the independent team of statisticians for analyses and results.
Enrolment was limited to men and women between the ages of 18 and 55 years with the diagnosis of RRMS according to the revised 2010 McDonald criteria38; who had a score of 0.0–5.0 in the Expanded Disability Status Scale (EDSS), with higher scores indicating more severe disease; who had undergone MRI showing lesions consistent with MS; who had had at least one medically documented relapse within the 18 months before their recruitment in this study and who had been receiving IFN-β treatment at last for the last 6 continuous months. If a clinical documented relapse was reported during the normalisation period, the entry baseline EDSS for that patient was considered as the EDSS Score documented at least 4 weeks after the last relapse during that period. Exclusion criteria were the use of prior immunosuppressant or monoclonal antibodies therapy, use of cytokine therapy in the last 3 months prior to randomisation, glatiramer acetate or intravenous immunoglobulins, or concomitant use of these treatments, pregnancy or nursing, any severe disease other than MS compromising organ function, a clinically significant infectious illness within 30 days prior to randomisation, history of recent drug or alcohol abuse, history of severe allergic or anaphylactic reactions or known specific nutritional hypersensitivity, consumption of any additional food supplement formula (prior use in the 3 months preceding randomisation, of any type of vitamin including vitamin D, or 6 months preceding randomisation, of any form of PUFA, or concomitant use of these treatments), prior or concomitant use of statins. Patients with primary progressive or secondary progressive disease were also excluded.
It was strongly suggested for the patients to continue only on the IFN-β treatment during the study duration. If a patient had to change disease-modifying treatment, then he/she was considered as a dropout, but continued to be medically followed.
Consumption of any additional food supplement formula, vitamin of any type or any form of PUFA supplement at any time during the trial was a reason for permanent discontinuation from the study. There were no other dietary intake restrictions. All dropouts continued to be medically followed and were strongly encouraged to remain in the study for follow-up assessments even if they had discontinued the assigned intervention formula. The intention-to-treat (ITT) population was defined as all randomised patients who had available data. The per-protocol population, used as a sensitivity analysis, was defined as all randomised patients who completed the follow-up without deviations from the protocol. All patients who transitioned from RRMS to secondary progressive MS, during the study period, were also excluded by protocol from the analysis to eliminate dramatic changes of increasing disability without any relapses.
Interventions
The daily oral dose of Neuroaspis plp10 dietary supplement was 20 mL containing a cocktail mixture of EPA (about 1650 mg)/DHA (about 4650 mg)/GLA (about 2000 mg)/LA (about 3850 mg)/total other omega-3 (about 600 mg)/total monounsaturated fatty acids (about 1700 mg)+total saturated fatty acids (18:0 about 160 mg, 16:0 about 650 mg)/vitamin A (about 0.6 mg)/vitamin E (about 22 mg) and pure γ-tocopherol (760 mg). Subjects were requested to consume supplement or placebo half hour before dinner.
The placebo was composed of pure virgin olive oil and was identical in colour, smell, shape, size and package to the Neuroaspis plp10 and both bottled in dark bottles under nitrogen bed. Both interventions contained food-grade citrus aroma (~3.5 mL) for palatability and taste reasons. The bottles were labelled with medication code numbers that were unidentifiable for patients as well as investigators. Both Neuroaspis plp10 and placebo were manufactured in Greece.
Study design, randomisation and blinding
The whole procedure followed the clinical trial guidelines as required by the Food and Drug Administration, according to the standards of the International Conference on Harmonization and the Committee for Medicinal Products for Human Use. The Clinical study was in agreement with the rules of Good Clinical Practice.
Subjects were randomly allocated into two groups per side to take either 20 mL dose of Neuroaspis plp10, dietary supplement formula or placebo for 30 months. Separate random scheme adopted for each centre (ie, randomisation stratified by centre). Randomisation was conducted by a computer-generated procedure that contained the two treatments in 1:1 ratio. Patients were allocated to the treatments according to the screening number ascending order.
The first 6 months were used as a normalisation period according to the protocol and previously published trial.39 This 6-month normalisation period would allow the interventions to exert their beneficial effect as oral PUFAs need 4–6 months to achieve pivotal action on immune and neural cells, correction of antioxidant deficiencies and body PUFA redistribution, and an optimal normalisation of the EPA and DHA ratios.28 39 41 42 The study begun with patient’s enrolment on January 2016 and it was completed on 31 December 2018. After the 6-month ‘normalisation period’, there was a 24-month ‘on treatment’ period, whose beginning was considered as the baseline.
Subjects were requested not to change their ordinary physical activity or dietary habits during the trial. All study personnel involved in the conduct of the study as well as the statisticians and the investigators were unaware of the treatment assignments throughout the study. Treating/examining physician and patients were blinded to treatment allocation.
The interventions were consumed orally every day, 30 min before dinner by a dosage calibrated cup continuously for 30 months. All participants were receiving a reminder at their cell phones every day at 18:00 hours. The adherence to the Neuroaspis plp10 treatment was further followed by asking patients to return the empty medication containers in order to be replaced on the appropriate preassigned date according to each patients’ stock (the supply was enough for 3-month consumption for each patient). The compliance to the IFN treatment was monitored and ensured internally by the patients’ follow-up system available in each one of the involved sites. Blood samples were collected from all enrolled patients for routine haematological and biochemical blood tests at enrolment, baseline, 6, 12, 18 and 24 months.
The interventions were used as supplements and adjuvant therapies. Depending on the enrolled patient’s clinical status and in accordance with common practice, they continued to receive their indicated regular treatment, with persistent evaluation for any side effects and adverse events.
Study procedures and end points
At each study site, one examining neurologist was designated along with one substitute for special occasions, when needed. Examining neurologists performed objective evaluation with the use of the EDSS. The same physician, as best as possible, maintained the role of treating neurologist for a given subject throughout the study and the same person maintained the role of examining neurologist for a given subject throughout the study. The same neurologists were responsible for all aspects of patient care, including the management of adverse events and the treatment of relapses. The involved neurologists were experienced, trained to standardise EDSS scoring procedures and a common approach has been discussed and agreed over a teleconference meeting. The physicians were not in contact with patients in any other capacity.
Neurological and clinical assessments were scheduled at enrolment, baseline and every 6 months on treatment. The patients were also examined by the assigned neurologists at unscheduled visits within 48 hours after the onset of new or recurrent neurological symptoms (table 1).
Operation table scheme
At 2 years ‘on treatment’, primary end point was the ARR. A relapse was defined as new or recurrent neurological symptoms not associated with fever or infection that lasted for at least 24 hours and was accompanied by new neurological signs or worsening of pre-existing symptoms (that had been stable for at least 1 month). To constitute a confirmed relapse, the symptoms should be accompanied by an increase of at least half a point in the EDSS Score, of one point in at least two EDSS functional system scores (FSS) or of two points in one EDSS FSS. The annualised relapse rate for each treatment group was calculated as the total number of confirmed relapses divided by the total number of the days on study, multiplied by 365.25. Relapses were treated with methylprednisolone at a dose of 1 g intravenous per day for 3 days, followed by prednisone orally at a dose of 1 mg/kg of weight per day on a tapering scheme for 3 weeks, according to the protocol. The key secondary end point was the time to confirmed disability progression, defined as an increase of 1.0 point on the EDSS from a baseline score of 1.0 or more or an increase of 1.5 or more from a baseline score of 0.0, confirmed after 6 months, with an absence of an ongoing relapse at the time of assessment and with all EDSS scores measured during that time meeting the criteria for disability progression. The final EDSS Score was confirmed 6 months after the end of the study. Another secondary end point was the number of T1-enhancing lesions, and the number of new/enlarged hyperintense lesions on T2-weighted MRI of the brain scans at the end of the study in comparison to the corresponding ones from the enrolment period. The neurologists were responsible for the review of any adverse or side effects, examined patients and made all medical decisions. Patients were able to contact their corresponding neurologist at any time if there was any adverse event, side effect or allergic reaction.
MRI had been performed locally at the participating centres on 1.5 T scanners according to a prespecified protocol provided by the central reading facility (neuroimaging research unit, division of neuroscience, IRCCS San Raffaele Scientific Institute, Milan, Italy). MRI parameters were assessed (using central reading) at enrolment and at 30 months (end of study). The identification of white matter lesions was performed by consensus of two experienced observers, unaware of the assigned treatment and the volume of the identified lesions was measured using a semiautomated segmentation technique based on local thresholding (Jim V.7.0; Xinapse System, Colchester, UK). The following MRI measures were produced: number of T1-enhancing lesions at baseline and at end of study, T1-enhancing lesion volume at baseline and at end of study; cumulative number of new/enlarging T2-hyperintense lesions at end of study versus baseline; and T2-hyperintense lesion volume at baseline and at end of study.
Serious adverse events were defined as those resulted in admission to hospital, cause prolonged disability or death, or judged as life-threatening or otherwise medically significant.
Statistical analysis
The p values that are reported for most baseline demographic and disease characteristics were calculated by the Kruskal-Wallis H test. The unadjusted relapse rate was calculated as the total number of relapses divided by the total number of patient-years followed for each treatment group. Poisson regression was the main statistical method performed for the analysis of relapses and for the analyses of the T1 and T2 brain lesions. A negative binomial regression was also used as a sensitivity analysis. Analyses were adjusted for: age, sex, years of diagnosis, baseline EDSS and relapses during normalisation period. Log-rank tests and Cox regression were used for the analysis of the proportion progressing (Kaplan-Meier) by analysis of the time until the onset of the progression of disability that was sustained over 24 weeks.
Dropouts
Dropouts at any time, including those subjects that never received the assigned interventions, were followed like all other participants.
Missing data handling
All patients who prematurely discontinued the study drug were encouraged to continue in the study until the end, regardless of the treatments received. ITT analyses included all randomised patients who had available information. Imputation of missing data was based on the last observation carried forwards’ approach.
Patient and public involvement
Different studies with the specific intervention have been performed and completed, including a proof-of-concept trial resulting in different published papers. Results and data disseminated through brought media presentations, press releases, websites and with the patients, community in general, and scientists be informed and aware. Moreover, different patients’ forums were discussing the present clinical trial when enrolment was initialised promoting participation.
Results
Study population
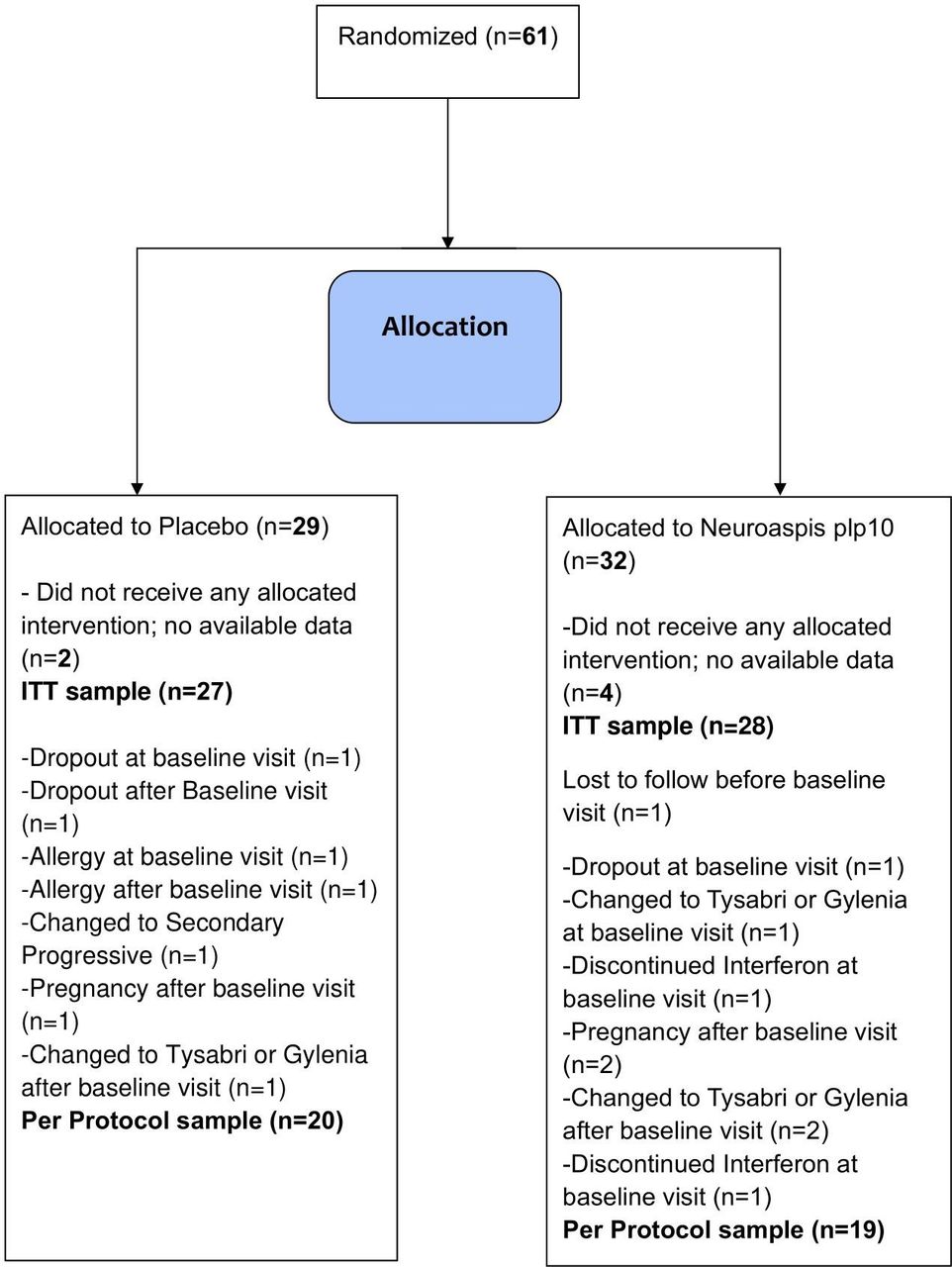
This is a phase III, double-blind, randomised, placebo-controlled clinical trial study that specifies definite clinical end points, in an attempt to demonstrate possible adjuvant therapeutic effects on disease-modifying therapy (DMT) in MS by a specific dietary/nutrition formula as previously described. Among the 61 patients, 32 patients were randomly assigned to receive the dietary formula and 29 to receive placebo (figure 1). There were no significant differences in baseline characteristics between the treatment groups for the ITT (table 2) or the PP (online supplemental table 1) populations. Six patients were lost to follow without ever been enrolled in the study and never consumed any of the interventions. Three patients dropped out before baseline and one patient before the 6 months on treatment. A total of 4 (6.5%) patients either withdrew consent (dropout) or lost in follow-up.
Supplemental material
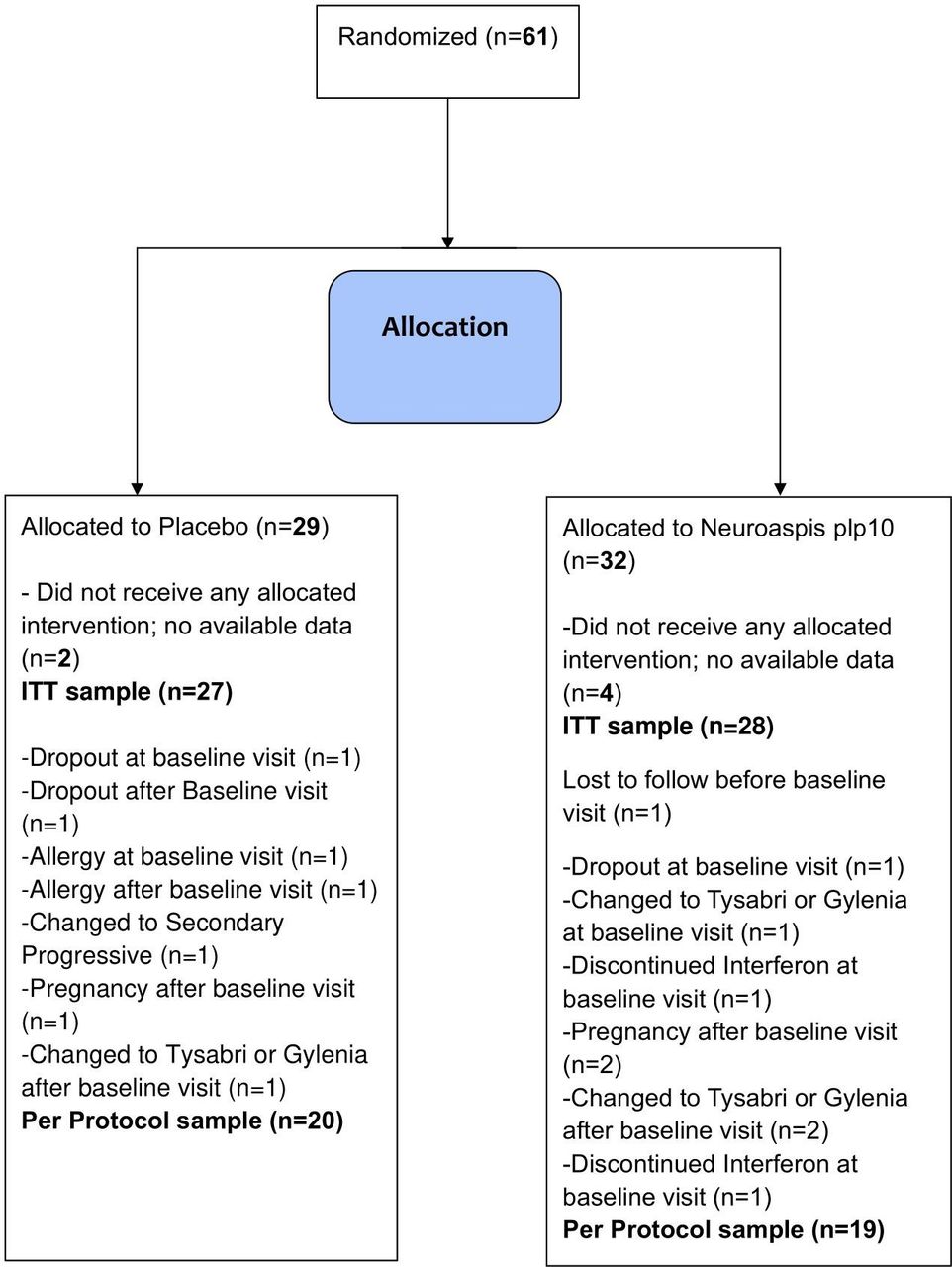
Trial profile. ITT, intention-to-treat.
Demographic, clinical and MRI baseline characteristics for the total randomised population (intention-to-treat (ITT) population) by treatment arm
Efficacy
Relapse
In the ITT analyses, after 1 year of treatment, Neuroaspis plp10 reduced the ARR to 0.13 (95% CI: 0.44 to 0.89) relapse per year, as compared with 0.63 (95% CI: 0.05 to 0.26) relapse per year in the placebo group with a corresponding 80% relative reduction in the annualised rate of relapse produced by Neuroaspis plp10 that was maintained at 2 years (Relative rate reduction (RRR), 0.2; 95% CI: 0.09 to 0.45; p<0.001) (the 2-year primary end point) (table 3). Analyses with adjustments for age, sex, years of diagnosis, baseline EDSS and relapses during normalisation period showed results consistent with the primary analysis. In the per protocol (PP) sensitivity analysis, Neuroaspis plp10 reduced the ARR to 0.00 (95% CI: 0.00 to 0.10; relapse free) as compared with 0.68 (95% CI: 0.45 to 0.99) relapses per year in the placebo group (p<0.001) and it was maintained at 2 years (online supplemental table 2).
Primary end point as determined by clinical results for the intention-to-treat (ITT) population
EDSS progression
A sustained progression of disability over 2 years (the 2-year secondary end point) was significantly less likely in the Neuroaspis plp10 group than in the placebo group (figure 2). At 2 years, the cumulative probability of progression (on the basis of Kaplan-Meier analysis) was 18.8% in the Neuroaspis plp10 group and 47.4% in the placebo group (HR, 0.27; 95% CI: 0.09 to 0.83; p=0.022), which represents a decrease of 28.6 percentage points or a relative 73% decrease in the risk of a sustained progression of disability with Neuroaspis plp10. In the (PP) sensitivity analyses, at 2 years, all patients in Neuroaspis plp10 group indicated no disability progression activity on the EDSS compared with the 55% cumulative probability of progression of the control group (online supplemental file 1).

{kind=link}
{kind=link}
Secondary end point, Expanded Disability Status Scale (EDSS) data analyses for the intention-to-treat population. Kaplan-Meier plots of the time to sustained progression of disability among patients receiving Neuroaspis plp10 as compared with placebo. Neuroaspis plp10 reduced the risk of sustained progression of disability by 73% over 2 years (HR, 0.27; 95% CI: 0.09 to 0.83). The cumulative probability of progression was 18.8% in the Neuroaspis plp10 and 47.4% in the placebo group.
MRI analysis
For ITT, Neuroaspis plp10 reduced the mean number of new/enlarging T2-hyperintense lesions over 30 months by 51% (incidence rate ratio (IRR), 0.49; 95%CI: 0.38 to 0.62; p<0.0001) as compared with placebo (table 4). Neuroaspis plp10 reduced the mean number of Gd-enhancing lesions by 71% (IRR, 0.29; 95% CI: 0.11 to 0.76; p=0.011) as compared with placebo at 30 months (table 4). Similar results were observed for the PP sensitivity analyses for both new/enlarging T2-hyperintense lesions (0.29; 95% CI: 0.20 to 0.41; p<0.0001) and Gd-enhancing lesions (0.29; 95% CI: 0.08 to 1.00; p=0.05) (online supplemental table 3).
Secondary end point as determined by MRI evaluation for the intention-to-treat population*
Safety
Over the course of the 30-month study, no severe adverse events were reported.
Discussion
MS is a chronic inflammatory multifactorial disease, where several different biochemical processes are simultaneously triggered and operating for the pathogenesis. Therefore, a much more dynamic and spherical approach has to be applied for simultaneous interference with all involved mechanisms and pathways that may potentially result to a long and effective treatment, possibly through the concept of the so-called ‘nutritional systems biology’.39 On the other hand, in a clinical trial setting in particular, the outcome should consider reliable surrogate measure sensitive to disease evolution such that it provides an answer on therapeutic effect quickly and in small numbers of subjects. Most importantly, it should reflect and predict an important clinical outcome. In MS, this outcome is either of the following: (1) reduction in relapse rate and/or (2) reducing the accumulation of irreversible disability.43
The aim of the Magnetic Resonance Image of Nutraceutical Efficacy on Relapsing-MS Autoimmune Lesions (MINERAL) Study was to investigate the increased efficacy due to the Neuroaspis plp10 dietary formula when systematically administered as adjuvant therapy to IFN, primarily on the relapse rate and secondary on the progression of irreversible neurological disability associated with relapsing MS and on the change in the number of MS-related brain lesions. No other published data for such an adjuvant efficacy study for comparable parameters were available. The results of the study support the hypothesis that Neuroaspis plp10, a cocktail mixture intervention of specific structural molecules, the omega-3 DHA and EPA, the omega-6 LA and GLA and the antioxidant proteins specifically vitamin E (α-tocopherol) and pure γ-tocopherol in a specific high molecular concentration can effectively contribute to the control of RRMS pathogenesis and progression. This can further support the hypothesis that all afore-mentioned molecules are possibly able to holistically interact on all levels of their side-of-action capabilities, including mostly the process of inflammation through promotion of the release of anti-inflammatory cytokines and the control of the release of proinflammatory cytokines by inhibiting the action of the inflammatory initiator molecule, the AA, an omega-6 PUFA and by quenching the overexpressed free radicals (hydro and nitro oxides) produced by the disease ongoing uncontrolled process of inflammation.39 As a result, the disruption of these processes and the resultant attenuation of inflammation may be beneficial to patients.
In patients with RRMS, Neuroaspis plp10 significantly reduced the risk of progression of disability and the ARR over 2 years of treatment. The effect of Neuroaspis plp10 was profound within 6 months after consumption and was sustained. In addition, efficacy was observed in terms of all secondary end points (73% reduction in EDSS for disease risk of disability progression and 71% reduction in incidence rate of lesions as detected by T2-weighted MRI and a 51% reduction in incidence rate of lesions as detected by T1-Gd-enhanced MRI) and all sensitivity analyses of the primary end points, indicating the robustness of the result.
DMTs have become the cornerstone of treatment for patients with RRMS. The IFN-β products and glatiramer acetate have shown that these agents reduce the annualised rate of relapse by about one third.44–47 In addition, neither IFN-β-1b nor glatiramer acetate had statistically significant effects on the progression of disability in patients with relapsing disease.45 46 All new drugs available for the treatment of RRMS are well studied and associated with a considerable means of effectiveness on both relapses, disability progression and brain lesions evolution, but are associated with considerable side effect.47–49 But, up to now, there is no intervention classified as food supplement and or as nutraceutical formulation that could be used effectively as an adjuvant to the existing RRMS treatments. The results of this study suggest that Neuroaspis plp10 may offer greater benefit to patients with RRMS when used as an adjuvant to the existing RRMS treatments.
The Neuroaspis plp10 formulation ingredients support completely different mechanisms of action than any of the currently approved RRMS treatments. This formulation may constitute a totally new approach in MS management probably by synergistically and simultaneously interfering on various major mechanisms and metabolic pathways involved in MS-related CNS inflammation, pathogenesis and disease evolution.
With much degree of a welcoming surprise for a second time (beside the previous reported proof of concept clinical trial findings), we have reached the conclusion that with this novel oral treatment formulation, associated with no significant side effects, as adjuvant to the previously considered ‘first-line’ drugs such as IFN-1b or similar (ie, glatiramer acetate) and possibly with the rest of the newer medications, we can introduce a safe and increased treatment effect instead of the reported efficacy of the DMTs alone.39
Several nutritional interventions have lately been considered as possible methods contributing to the quality of life of patients experiencing chronic neurological conditions.39 50 Similarly, Neuroaspis plp10 consists of a formulation with biophysical characteristics that can be considered as a medical nutrient concoction (nutraceutical) potentially for MS prevention and adjuvant treatment.
To conclude, the present study supports a link between, dietary, nutritional, immunological and inflammatory aspects of MS and identifies an important new potential horizon in the approach of MS prevention and treatment. With limitation, the small number of patients in the trial, the length of the study, as well as the multiplicity of the analyses and the solid protocol can all be considered as positive parameters for the robustness of the final indication and conclusion.
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Cyprus national bioethics committee; national ethics committee of Greece, ID: EEBK/EΠ/2013/18. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank George Miltiadous, MD, Hippocrateon Private Hospital, Nicosia, Cyprus for his outstanding contribution to the project as the independent control and safety committee. An abstract based on this clinical trial project was presented in the 9th IMBMC Congress, 18–20 November 2021.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @paolopreziosa
MCP and IP contributed equally.
Contributors MCP and IP were responsible for the protocol and study design. PV, RM, PP and MAR were responsible for the MRI protocol and MRI data analyses. IP was the author responsible for the overall content as the guarantor. EN and GM statistically analysed the collected data. MCP, CB, NG, GH and ED were the evaluators of patients’ clinical progress signs and the treating physicians. EN, GM, MCP, RM, PP, MAR and IP interpreted the data. IP, GL and SO were responsible for the laboratory work. MCP and IP wrote the article. All authors contributed to the editing of the manuscript and vouch for the accuracy and completeness of the data and the statistical analysis.
Funding This study was funded by Cyprus Ministry of Energy, Commerce, Industry and Tourism—Structural Funds Program (8.1.12.13.2.3.60).
Competing interests MCP, GL, IP received grand support from the Cyprus Ministry of Energy, Commerce, Industry and Tourism—Structural Funds Program (Creation of New High Technology and Innovation Enterprises). PALUPA Medical is a research company formed and registered under the Incubator program. MCP, GL and IP are stockholders of PALUPA Medical. MAR received speaker honoraria from Bayer, Biogen, Bristol Myers Squibb, Celgene, Genzyme, Merck Serono, Novartis, Roche and Teva, and receives research support from the MS Society of Canada and Fondazione Italiana Sclerosi Multipla. NG received speaker honoraria from Bayer, Biogen, Gensis Pharma, Celgene, Sanofi, Merck, Novartis, Roche and Teva, and receives research support from Biogen and TEVA. CB received travel support and/or lecture fees from Novartis, Merck, Genesis, Sanofi, Teva, Roche and Mylan. GH, ED, EN, GM, SO, PV, RM and PP declare no competing interests. No pharmaceutical companies were involved in this project. The intervention, trade marked as Neuroaspis plp10, is under a US patent application, and covered with granted patent in several other countries including Canada, Japan, China, Ukraine, Australia, Israel and New Zealand.
Provenance and peer review Not commissioned; internally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.