Article Text

Abstract
Introduction Mitochondrial diseases exhibit wide phenotypic heterogeneity, and can present as progressive myoclonic epilepsy.
Summary We report a case of adult-onset drug-resistant epilepsy, cortical myoclonus and bilateral optic neuropathies due to m.14487T>C, a rare mitochondrial gene mutation identified on whole-genome sequencing. This mutation, which affects the NADH dehydrogenase 6 (ND6) subunit of the mitochondrial respiratory chain, is most commonly implicated in cases of infantile-onset Leigh syndrome, although a broader phenotypic spectrum including migraine with aura and progressive myoclonic epilepsy have been described. Serial MRI scans over a 2-year period demonstrated the interval development of bihemispheric stroke-like lesions. Giant somatosensory evoked potentials and short-duration myoclonic jerks with craniocaudal spread on surface electromyography were consistent with cortical myoclonus. Optical coherence tomography showed bilateral symmetric thinning of the nerve fibre layer in the papillomacular bundles.
Conclusion Whole-genome sequencing can help to provide a definitive diagnosis for mitochondrial disease and should be considered in situations where clinical suspicion remains high despite normal genetic panels or muscle histopathology. Mitochondrial disease can present as adult-onset progressive myoclonic epilepsy, and bilateral optic neuropathies can be a striking feature of ND6 mitochondrial gene mutations. In our case, severe cortical myoclonus affecting speech and swallowing remained highly drug-resistant, however, symptomatic benefit was derived from targeted onabotulinum toxin A injections.
- epilepsy
- mitochondrial disorders
- neurogenetics
- botulinum toxin
Data availability statement
All relevant data are included in the article.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Mitochondrial diseases can present with adult-onset progressive myoclonic epilepsy, an uncommon and genetically heterogeneous syndrome characterised by seizures, myoclonus and varying degrees of ataxia and dementia. A positive family history, other coexisting features such as polyneuropathy, optic neuropathy and stroke-like lesions in non-vascular territories can help suggest the diagnosis of mitochondrial disease, although there is much phenotypic variability. In cases where clinical suspicion is high but mitochondrial gene panels and muscle histopathology are non-diagnostic, mitochondrial whole-genome sequencing is emerging as an affordable and increasingly available method of establishing a definitive diagnosis.
Case report
A 43-year-old man was referred with a 4-year history of drug-resistant epilepsy characterised by focal to bilateral tonic-clonic seizures and refractory myoclonus. There was no other medical history. He had been born at term, attained all developmental milestones normally and had no history of childhood convulsions.
He had two sisters, both of whom had childhood epilepsy. One sister had experienced global developmental delay, had never talked and been wheelchair-bound following a diagnosis of congenital muscular dystrophy. The other sister had reached normal milestones until the age of 3 years, after which she developed progressive gait abnormalities. Both sisters had died from pneumonia aged 13 and 16 years old. He had one daughter, aged 8 years old, with no neurological symptoms (online supplemental figure 1).
Supplemental material
Our patient reported developing symptoms when he was 39 years old, initially with episodes of right elbow cramping, which evolved to paroxysmal episodes of right upper limb jerking lasting approximately thirty seconds on each occasion. With time, these attacks became more frequent, and he also began to experience episodes of twitching over the right side of his face. He reported violent episodes lasting up to fifteen minutes with jerking of all four limbs with preserved awareness. He had one episode associated with loss of consciousness, followed by right upper and lower limb weakness lasting several days. When he was 42 years old, he developed bilateral simultaneous progressive visual loss over a period of weeks, to the extent that he had been registered legally blind.
On examination, there was high-frequency, stimulus-sensitive multifocal myoclonus predominantly affecting the right side of his face and right upper limb. Awareness was preserved. Facial myoclonus grew more pronounced when talking to the extent he was unable to speak or swallow (online supplemental video). Ankle reflexes were absent and there was a glove-and-stocking distribution of sensory loss.
Supplementary video
Initial visual testing revealed bilateral central scotomata with corrected visual acuities of 6/60 in the right eye and 6/18 in the left eye, relatively preserved peripheral vision and pallor of both optic discs (figure 1). Optical coherence tomography showed bilateral symmetric thinning of the nerve fibre layer in the papillomacular bundles. Six months later, there had been considerable deterioration in central vision, with acuities in both eyes at the level of hand movements only. This did not improve despite a trial of high dose intravenous steroids with an oral taper.
{kind=link}
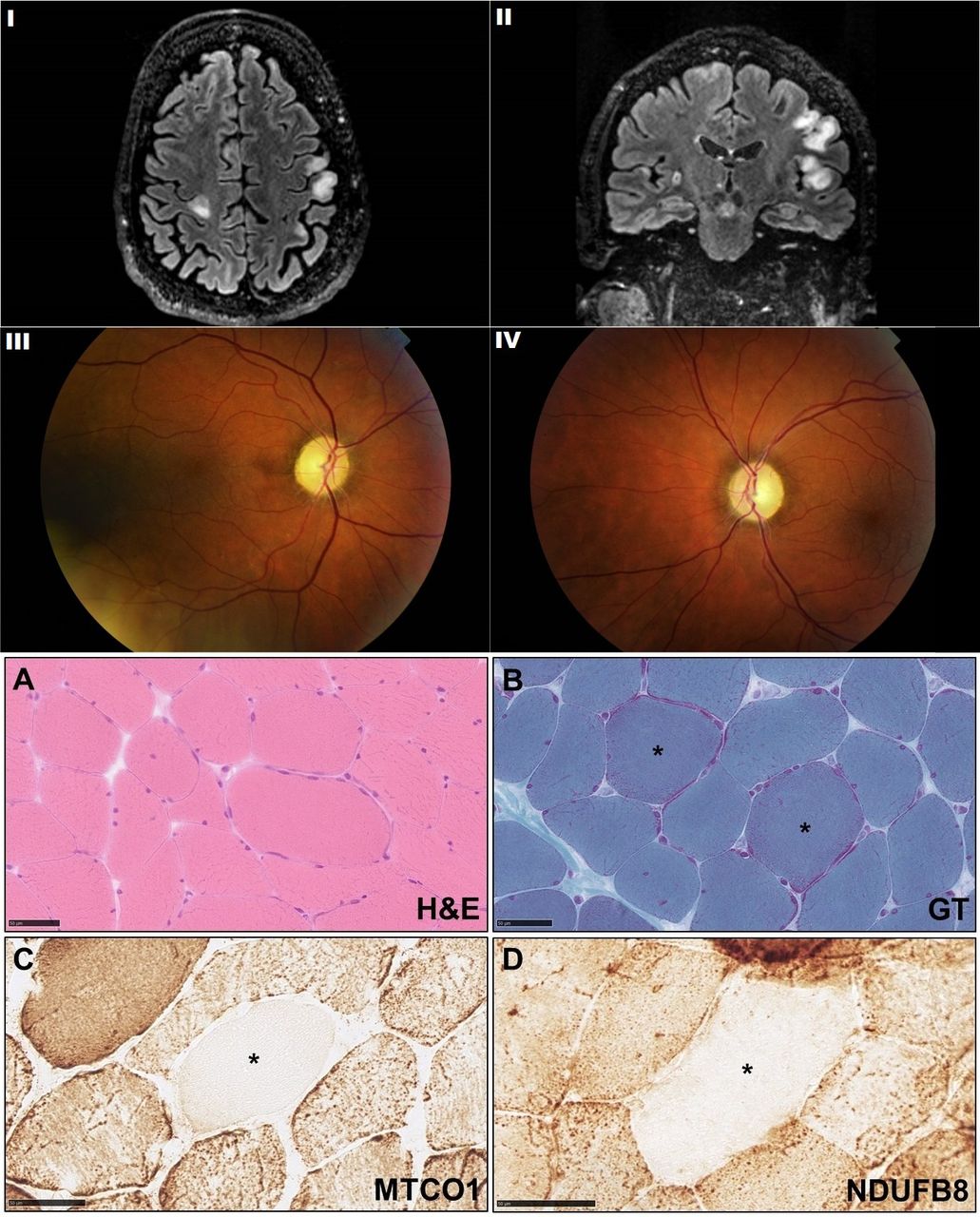
I, II: axial and coronal fluid attenuated inversion recovery (FLAIR) sequences on MRI brain demonstrating cortical hyperintensities involving both hemispheres. III, IV: right and left fundal photographs demonstrating pale optic discs consistent with optic atrophy. Muscle biopsy histopathology: (A) H&E demonstrating mild increase in variation of fibre size with occasional fibres containing internal nuclei; (B) rare fibres with prominent subsarcolemmal mitochondria were seen (asterisk) reminiscent of but not diagnostic of ragged red fibres; (C) a rare fibre with negative staining for complex IV component, MTCO1 has been identified (asterisk); (D) the complex I component immunohistochemistry also shows a rare fibre with very pale staining (asterisk). GT, Gomori Trichrome; MTCO1, mitochondrially encoded cytochrome c oxidase I (complex IV component); NDUFB8, NADH dehydrogenase (ubiquinone) 1 beta subcomplex subunit 8 (complex I component).
MRI scans over a 2-year period demonstrated interval development of multifocal cortical and subcortical T2/FLAIR hyperintensities involving both cerebral hemispheres (figure 1). Nerve conduction studies revealed a symmetrical, length-dependent axonal neuropathy. Prolonged video electroencephalography (EEG) revealed episodes of twitching involving the face, upper body and right arm with no epileptiform correlate. Although excessive muscle artefact made back-averaging impossible, short-duration myoclonic jerks with craniocaudal spread, followed by brief periods of atonia were consistent with cortical myoclonus. Giant somatosensory evoked potentials (N20-P25 24.4µV) were also found, supporting the presence of cortical hyperexcitability.
A muscle biopsy was performed, which showed minor non-specific changes (figure 1). Although there were no definite cytochrome oxidase negative fibres (which may be subject to variation in level of sections), rare fibres deficient in complex I and complex IV components were evident. These changes were not diagnostic, but were in keeping with the clinical impression of a mitochondrial disorder. On the contrary, muscle respiratory chain enzyme analysis showed no evidence of complex I/II+III/IV or ubiquinone deficiency. A 21-gene panel for pathogenic mitochondrial gene mutations was negative. Given clinical suspicion, we proceeded to whole mitochondrial gene sequencing on the muscle sample that revealed a 98% heteroplasmic m.14487T>C p. (Met63Val) mutation. This is a pathological variant which results in an amino acid substitution in NADH dehydrogenase 6 (ND6), a complex 1 subunit of the mitochondrial respiratory chain.
Management
A number of different antiseizure medications including levetiracetam, carbamazepine, zonisamide, perampanel and clonazepam (up to 2 mg four times per day) were trialled. These were associated with initial improvement in myoclonus, however, benefit was not sustained. Levetiracetam was tapered to cessation and replaced with piracetam (4 g three times per day), with mild benefit.
Videofluoroscopy revealed oropharyngeal myoclonus impacting on his ability to safely swallow and a percutaneous endoscopic gastrostomy was inserted to assist with nutritional demands.
Onabotulinum toxin A was administered to a number of right-sided facial, neck and upper limb muscles (80 units orbicularis oculi, 10 units nasalis, 10 units risorius, 20 units levator anguli oris, 60 units platysma) with a 70% subjective improvement in myoclonus (online supplemental video).
Discussion
This case is notable for a number of reasons. It illustrates how mitochondrial disease must always remain in the differential of an adult who presents with progressive neurological symptoms in the setting of a positive family history. Moreover, it demonstrates how uncovering the diagnosis of mitochondrial disease can be challenging, and ongoing persistence remains key to being able to establish a definitive diagnosis.
m.14487T>C is a rare pathogenic mitochondrial DNA (mtDNA) mutation that affects the ND6 subunit of respiratory chain complex 1, and was originally reported in isolated cases of infantile-onset Leigh syndrome.1 Patients with this mutation may exhibit other mitochondrial features such as cardiomyopathy or diabetes, and the severe, progressive visual impairment in our case was a striking feature.2 This phenotype, reminiscent of Leber’s hereditary optic neuropathy, has been seen in other mutations affecting ND6, although has only rarely been reported in the setting of m.14487T>C mutation.3 4
Although unconfirmed, it is likely our patient’s two sisters carried the same mutation, presenting with a Leigh-like phenotype. Although we could not review his sister’s muscle biopsy, this was reported as normal. Notably however, as in our case, muscle biopsy findings can show minor non-specific changes (or even be normal), highlighting the emerging paradigm that favours early genetic tests in cases of suspected mitochondrial disease.2 5 Full sequencing of mtDNA is becoming increasingly available and is particularly useful in this setting where clinical suspicion is high but histopathology and genetic panels are non-diagnostic. A definitive diagnosis was very helpful in our patient, allowing us to counsel that risk to his daughter was negligible, informing prognosis and opening up potential research eligibility.
This case also underscores the continuum between seizures and cortical myoclonus, both of which are driven by abnormal neuronal hyperexcitability. Absence of electrographic findings on scalp EEG during clinical episodes may raise a question regarding a functional aetiology. As in this circumstance, multi-channel time-locked EEG was essential in diagnosing cortical myoclonus. This was further confirmed through the demonstration of giant somatosensory evoked potentials.
While onabotulinum toxin A has been more commonly used for the treatment of peripheral myoclonus such as hemifacial spasm, we describe its successful use as a palliative remedy for refractory cortical myoclonus. This treatment was necessary as he remained markedly symptomatic despite use of five simultaneous antiseizure medications (levetiracetam, perampanel, zonisamide, carbamazepine and clonazepam). As is our usual practice in mitochondrial disease, we avoided the use of sodium valproate, which can be associated with fulminant hepatic failure as a consequence of carnitine deficiency.
A multidisciplinary approach was essential, particularly in light of his clinical progression. Mindfulness was introduced as an effective strategy, and he continues to receive input from clinical psychology and rehabilitation services. Although referred for hospice care, we have illustrated how a range of symptomatic therapies to assist with speaking, swallowing, nutrition and the management of refractory myoclonus can help maximise quality of life in patients with progressive myoclonic epilepsy secondary to mitochondrial disease.
Data availability statement
All relevant data are included in the article.
Ethics statements
Acknowledgments
MKS is supported by the UCLH Biomedical Research Centre.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
AK and SN are joint first authors.
Contributors AK, SN and SBW wrote the first draft. AM, FB and MKS made critical revisions for intellectual content. All authors approved the manuscript for submission.
Funding The article processing charge was funded by UCL.
Competing interests None declared.
Provenance and peer review Not commissioned; internally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.