Article Text

Abstract
Objective To present a case of two identical twins presenting concurrently with symptoms and subsequent initial diagnosis of neuromyelitis optica spectrum disorder (NMOSD).
Methods Clinical, laboratory and MRI findings for both twins were reviewed and presented here.
Results Twin A presented with right eye pain and subsequent blurred vision in right eye. MRI of the brain and spine demonstrated pre-chiasmal right optic nerve enhancement and T2 hyperintense lesions in the spinal cord at T7 and T9 levels. Cerebrospinal fluid (CSF) analysis was remarkable for NMO/aquaporin-4 (AQP4) fluorescence-activated cell sorting (FACS) titre of 1:32 and a serum NMO/AQP4-IgG positive titre of 1:10 000. Twin B presented with diplopia. MRI of the brain and spine demonstrated T2 hyperintense lesions in the periventricular cerebral white matter, in the periaqueductal white matter of the pons, in the midbrain and the cervical spinal cord. Neurological examination findings revealed incomplete right trochlear palsy, rotatory nystagmus, an incomplete left internuclear ophthalmoplegia and hyper-reflexia. CSF analysis was remarkable for NMO/AQP4 FACS titre of 1:256 and a serum NMO-IgG positive titre of 1:10 000. Both twins responded well to intravenous steroid therapy. There was no adverse environmental exposure present.
Conclusion We present an interesting and rare case of identical twins presenting concurrently and for the first time with NMOSD.
- neuroimmunology
- neuro-ophthalmology
- neuropharmacology
- neuroradiology
- multiple sclerosis
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study. All data are available (images and laboratory results) and submitted in this manuscript.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Case report
We describe identical twin sisters with no known family history of demyelinating disease, including neuromyelitis optica spectrum disorder (NMOSD). Their mother and maternal grandmother have Leiden factor V mutation. They have no knowledge of their father’s medical history.
Patient A is a 20-year-old right-handed African-American woman and the bigger of the twins. All of her developmental milestones were met. She has hypothyroidism since the age of 9 years, irritable bowel syndrome, vitamin D deficiency and factor V Leiden homozygous. Two weeks prior to her clinic visit, she developed an insidious onset of right eye pain with eye movement. Nine days later, she developed blurred vision in the right eye only. She denied diplopia, headaches, fever, COVID-19 exposure or any symptoms suggestive of viral infection.
Several hours prior to this clinical visit, she was evaluated by an ophthalmologist who felt the patient had acute right retrobulbar optic neuritis. Her neurological and physical examination was unremarkable. Her visual acuity was not assessed due to chemically dilated pupils.
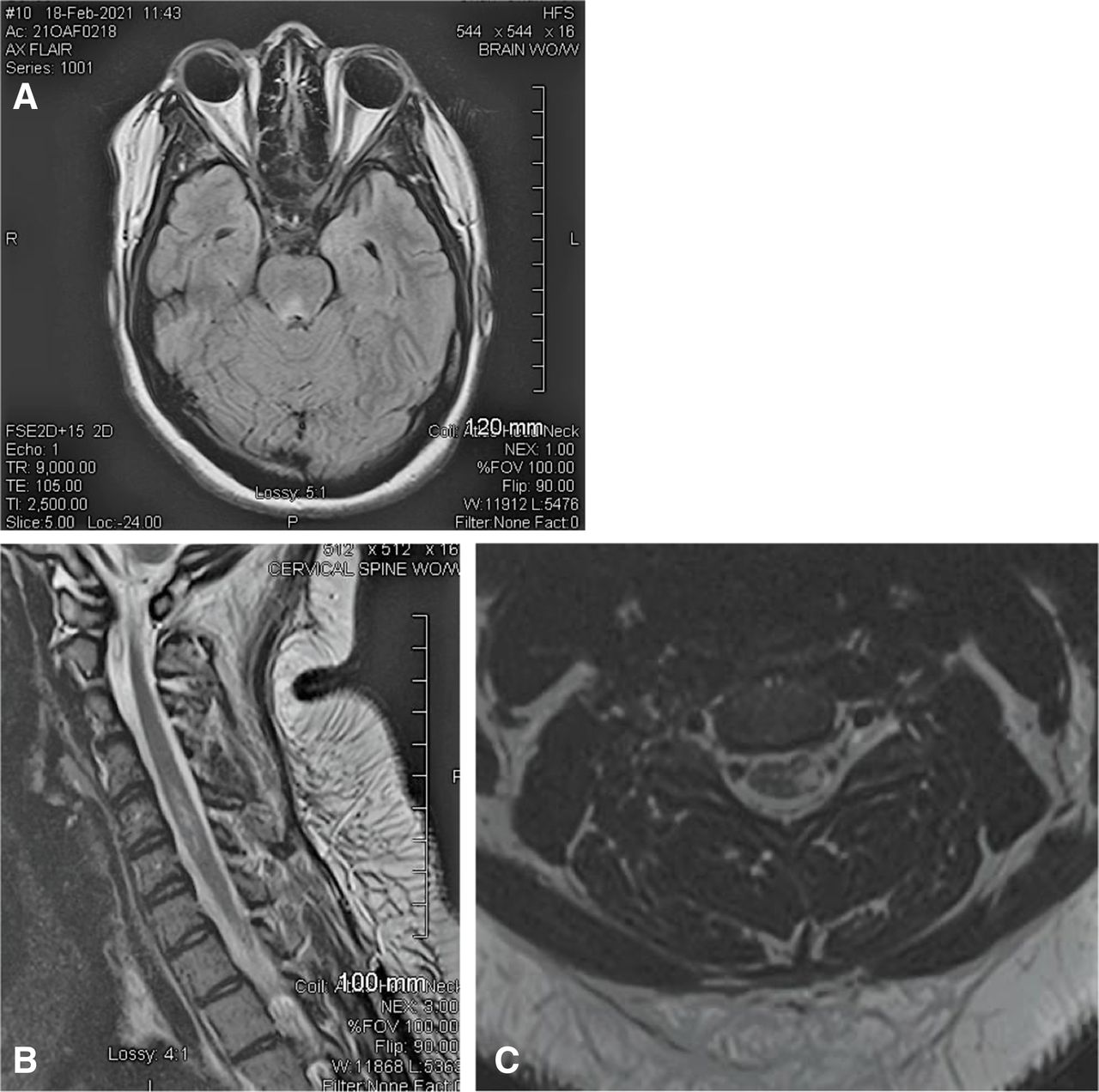
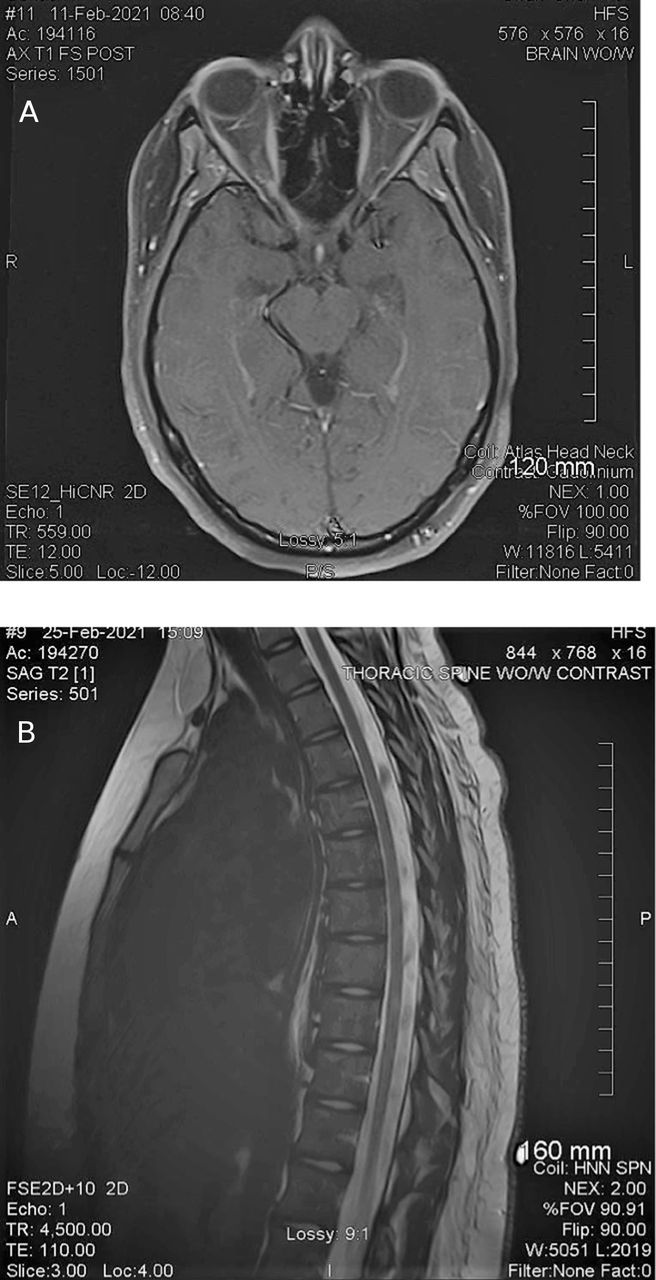
MRI of the brain was obtained the next day and demonstrated right optic nerve enhancement at the level of the orbital apex and immediately proximal to the optic chiasm (figure 1A). The lumbar puncture was done the same day and was remarkable for an opening pressure of 39 cm of water. Closing pressure was 19 cm of water. Cerebrospinal fluid (CSF) studies were remarkable for positive NMO/aquaporin-4 (AQP4), titre 1:32 and a serum NMO/AQP4-IgG positive titre of 1:10 000. CSF IgG index was 0.56 (reference range <0.85). CSF cultures were negative, including a negative Lyme test. ACE was 0.7 (reference range 0.0–2.5). No oligoclonal bands. No red and three white blood cells. She had normal protein and glucose. Cytology was normal. Metabolic work-up was normal.
(A) Patient A—MRI of the brain. (Left) Patient A—right optic nerve signal abnormality consistent with optic neuritis. (B) MRI of thoracic spine reveals two small foci of increased T2 signal within thoracic cord at the T7 and T9 levels.
MRIs of cervical and thoracic spine were performed 2 weeks later. MRI of thoracic spine revealed two small foci of increased T2 signal within thoracic cord at the T7 and T9 levels without contrast enhancement (figure 1B). MRI of cervical spine and spinal cord was normal (not shown).
The patient subsequently underwent intravenous methylprednisolone treatment (1 g daily for 5 days followed by a prednisone taper). Her vision returned to normal after 18 days and the remainder of her neurological examination at that time was also normal. There was no adverse environmental exposure present.
Patient B is a 20-year-old right-handed African-American woman. All developmental milestones were met. She has hypothyroidism and factor V Leiden homozygous. She denied any history of neurological symptoms suggestive of demyelinating disease.
Six weeks before her neurological evaluation, she began seeing ‘things’ before falling asleep. Her primary doctor felt it may be related to a urinary tract infection. She was started on antibiotics and the visual symptoms resolved after several days. She remained asymptomatic until 3 days prior to her neurological evaluation when she woke up with double vision. She denied any fever, headache, slurred speech, eye pain, blurred vision or any other symptoms. Her symptoms remained unchanged for 3 days. No exposure to COVID-19.
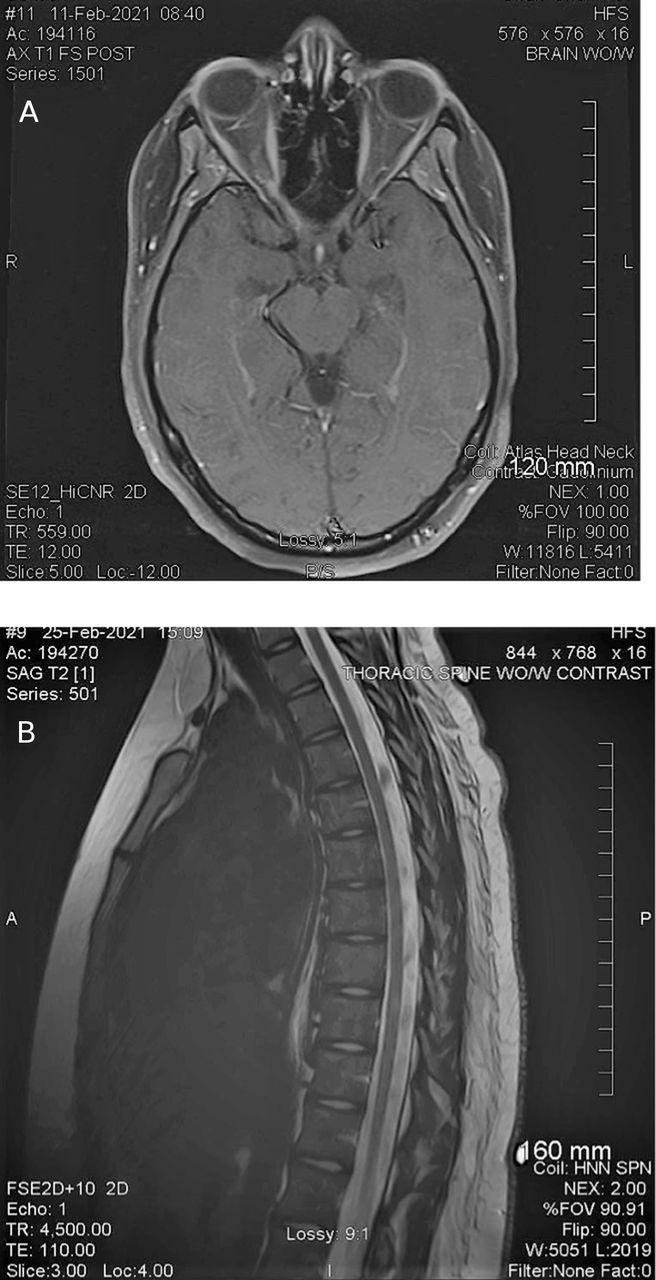
Metabolic work-up was remarkable for a thyroid-stimulating hormone of 0.05 (normal 0.3–5.0) and vitamin B6 deficiency. Brain MRI was performed on the day of her neurology appointment. It demonstrated on T2 and fluid-attenuated inversion recovery hyperintense signal abnormalities in the periaqueductal grey matter (figure 2A). MRI of cervical spine demonstrated non-enhancing T2 hyperintense signal abnormalities in the left hemi-cord at the levels of the C2, C5 and C6 vertebral body (figure 2B,C). MRI of thoracic spine was normal.
{kind=link}
{kind=link}
(A) Patient B—MRI of the brain; hyperintense signal abnormalities in the periaqueductal grey matter. (B,C) MRI of cervical spine (sagittal and axial images) non-enhancing hyperintense signal abnormalities in the left hemi-cord at the levels C2, C5 and C6 vertebral body.
Neurological and physical examinations were remarkable for incomplete right trochlear nerve palsy, rotatory nystagmus, an incomplete left internuclear ophthalmoplegia and hyper-reflexia without any pathological reflexes. The patient underwent a lumbar puncture remarkable for an opening pressure of 39 cm of water (the same as her sister) and closing pressure was 16 cm of water.
CSF studies revealed positive NMO/AQP4, 1:256 and a serum NMO/AQP4-IgG positive titre of 1:10 000. CSF IgG index was normal. Oligoclonal bands were 2, which were borderline. The remaining tests including ACE, Lyme, viral and bacterial cultures were negative.
Like her sister, the patient underwent intravenous methylprednisolone treatment followed by a prednisone taper. Twelve days following the initial examination (and start of treatment), the patient’s neurological examination was normal. There was no adverse environmental exposure present.
Background
NMOSDs are autoimmune inflammatory diseases affecting the central nervous system, predominantly characterised by optic neuritis and myelitis.1 The discovery of AQP4-specific IgG has expanded the understanding of NMOSD.2 Serum anti-AQP4 titres have been reported to correlate with disease severity, that is, decrease following treatment and stay low during remission.3
NMOSD is associated with autoimmune disorders including hypothyroidism, myasthenia gravis and pernicious anaemia.4 The prevalence of NMOSD in adults ranges from 0.37 to 10 per 100 000. It affects women 7.6 times more often than men, and there are geographical and ethnic differences in disease prevalence.5 NMOSD typically occurs sporadically although rare familial cases have been reported.6 7
Central nervous system involvement is not only limited to the optic nerves and the spinal cord, but can also affect the brainstem and diencephalon.
MRI lesions are found in regions of high AQP4 expression. It can cause encephalopathy, posterior reversible encephalopathy, hypothalamic dysfunction, myopathy, autonomic dysfunction and in rare instances, death from respiratory failure secondary to brainstem lesions. NMOSD is a relapsing disease in approximately 90% of affected individuals, and in 60% the second attack will occur within the first year after onset.8
Diagnostic criteria are based on core clinical findings, AQP4 results and MRI.9 The AQP4 water channel is the antigen of NMO-IgG and has a pivotal role in the pathogenesis of NMOSD. AQP4-IgG is a specific biomarker for NMOSD.10 The AQP4-IgG antibody testing should be performed prior to initiation of immunotherapy.10 CSF is often abnormal, including elevated protein and white cell count. Oligoclonal bands are absent in 70%–80% cases.10
Abnormalities of the spinal cord are seen on MRI in up to 60% of patients and consist of extensive lesions often involving three or more vertebral levels.11 The central grey matter (along the central canal) involvement in the spinal cord is common and corresponds to the prominent expression of AQP4 antigen in the spinal cord. Lesions typically involve a large cross-sectional area of the spinal cord. ‘Bright spotty lesions’ are lesions that are markedly T2 hyperintense (more intense than CSF) and have been described as a relatively specific finding in NMOSD.11 MRI of the brain (excluding the optic nerve abnormalities) is initially normal in 55%–84% of patients with NMOSD. MRI reportedly becomes abnormal in up to 85% cases.12
The acute attacks of NMOSD are treated with high-dose intravenous methylprednisolone or plasma exchange for patients unresponsive to steroid treatment. Long-term immunotherapy is recommended for the prevention of attacks as soon as the diagnosis of NMOSD is established.13
Discussion
NMOSDs are usually sporadic disorders. Familial form does occur in approximately 3% of patients and cannot be differentiated from sporadic form based on clinical presentation, gender, age at onset or frequency of NMO-IgG detection. This suggests a complex genetic predisposition for the disease. Ethnic predilection is controversial.14
Identical twin sisters (24 and 26 years of age at the onset of NMOSD symptoms) were reported in 1938. Both had severe episodes of transverse myelitis and bilateral optic neuritis and died of related complications. The autopsy revealed demyelination of the optic nerves and spinal cord.6 A study from Japan reported two sisters with early onset (age 2 and 3 years at onset) who presented with bilateral optic neuritis and thoracic myelitis and another case of two sisters with late onset of NMOSD (59 and 62 years of age).15
NMOSD is characterised by acute relapses in sporadic and familial series. Optic neuritis and myelitis are the dominant clinical presentations.4
This case is unique in that these identical twins presented essentially simultaneously with different clinical symptoms and different radiological findings, not matched in the literature since the original twin article in 1938 (they were 2 years apart).6 Both had the spinal cord involvement, had hypothyroidism, Leiden factor V homozygous and had no previous symptoms and, interestingly, the same opening pressure. Their AQP4 titre was disproportionately elevated in the twin with brainstem involvement, which corresponds with the size of the lesion.3 Both sisters had an excellent response and recovery after intravenous methylprednisolone treatment.
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study. All data are available (images and laboratory results) and submitted in this manuscript.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors IB contributed to planning, conduct and reporting. IB is also responsible for the overall content as a guarantor. JIH contributed to planning, conduct and reporting. MM contributed to planning, conduct and reporting. RJW contributed to planning, conduct and reporting.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.