Article Text

Abstract
Background Patients with ornithine transcarbamylase deficiency (OTCD) often present with severe hyperammonaemia. We report a case of osmotic demyelination syndrome (ODS) secondary to the treatment of hyperammonaemia due to OTCD, a disease requiring early diagnosis, as it can have a severe prognosis.
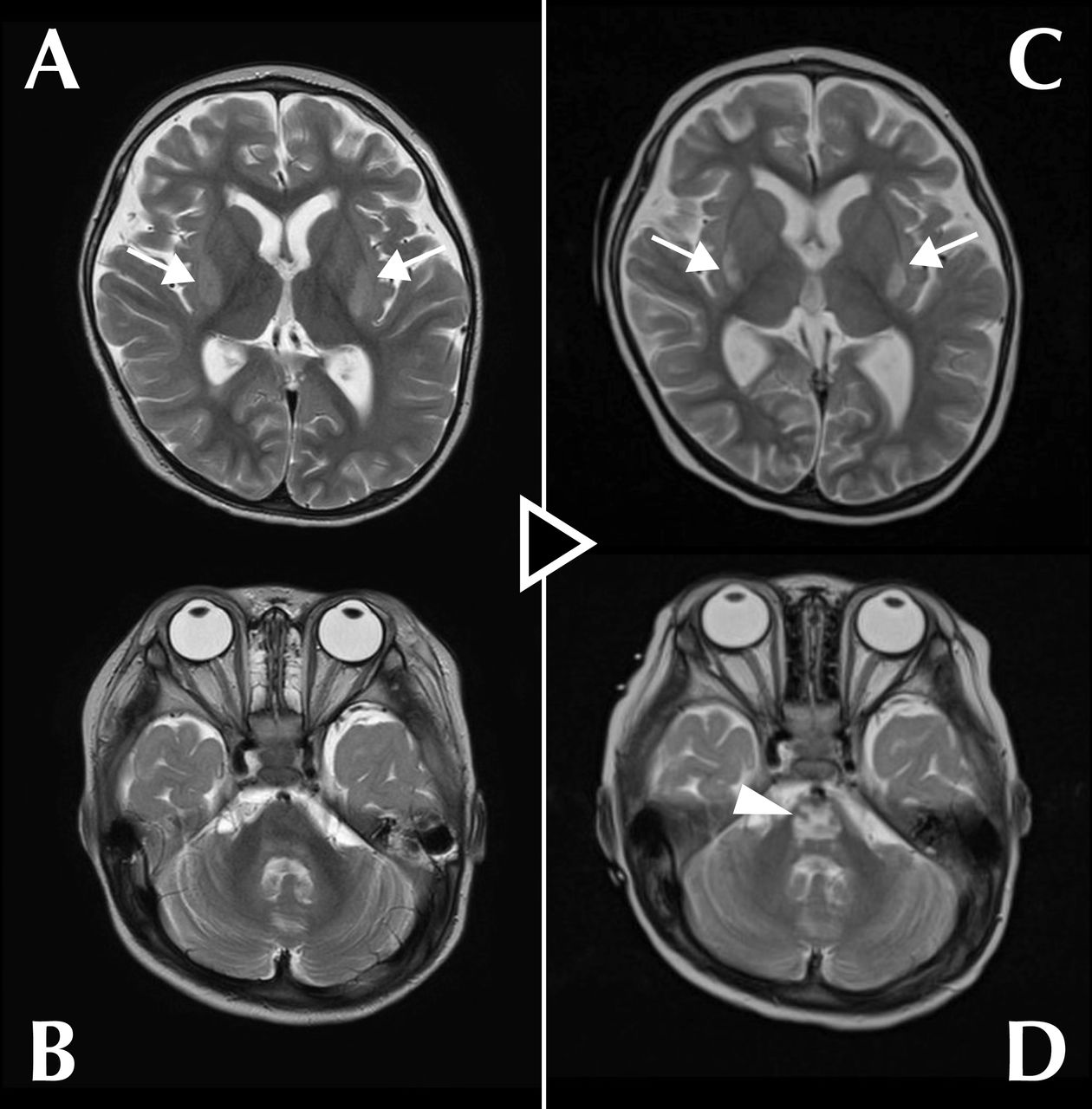
Case A girl toddler was brought to the hospital with a complaint of somnolence, presenting with hyperammonaemia and liver failure, and was diagnosed with OTCD. Treatment was started immediately, and the ammonia level returned to the normal range within 24 hours. On days 13–20, another treatment was commenced for re-elevated ammonia levels, which subsequently returned to within the reference range; however, mildly impaired consciousness persisted. Hypokalaemia coincided with temporary intravenous treatment and continuous haemodialysis. T2-weighted magnetic resonance images revealed lesions as high-signal areas in the bilateral putamen on day 11 (extrapontine myelinolysis (EPM)) and in the pons on day 51 (central pontine myelinolysis (CPM)). Consequently, ODS was diagnosed.
Conclusion When interpreting magnetic resonance images of patients under acute treatment for hyperammonaemia due to OTCD, a condition that may be complicated by hypokalaemia, paying attention to findings suggesting EPM may help detect ODS before CPM appears and may improve patient prognosis.
- MRI
- MYELOPATHY
- NEURORADIOLOGY
- PAEDIATRIC
- METABOLIC DISEASE
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Ornithine transcarbamylase deficiency (OTCD) is the most common urea cycle disorder. It is an X-linked genetic disorder caused by an abnormality in the ornithine transcarbamylase (OTC) gene on Xp21.1. Patients with OTCD have hyperammonaemia due to a defect in the OTC enzyme, a member of the urea cycle responsible for detoxifying ammonia. OTCD presents with various neurological symptoms.1
There are two reports of osmotic demyelination syndrome (ODS) observed after correcting hyperammonaemia due to OTCD.2 3 ODS is characterised by demyelination in the pontine centre, basal ganglia, thalamus and cerebellar white matter due to osmotic stress to cells caused by rapid changes in osmolyte concentrations.4 5 While well known for its development when treating hyponatraemia, ODS occurrence following hypokalaemia has also been noted,6–8 although less attention has been paid to this pathogenetic factor. ODS causes various neurological symptoms, some of which have serious consequences, including locked-in syndrome, coma and death,4 5 thus emphasising the need for early diagnosis.4 9 The gold standard diagnostic tool for ODS is MRI,4 which detects lesions in the centre of the pons, known as central pontine myelinolysis (CPM), and other areas such as the basal ganglia and thalamus, known as extrapontine myelinolysis (EPM).4 5 10 Recently, EPM has been suggested to appear before CPM in cases where both CPM and EPM are present, making EPM a potentially useful finding for early ODS detection.9 11
We report hyperammonaemia and hypokalaemia-associated ODS secondary to the treatment of hyperammonaemia in a patient with OTCD, in whom MRI captured the appearance of EPM before CPM. This report suggests that patients under acute care for OTCD might be prone to developing ODS. EPM identification may lead to a better prognosis for these patients by predicting and managing the subsequent emergence of CPM.
Case report
A girl toddler was brought to the hospital with the chief complaint of somnolence for approximately 3 months. Analysis of blood and urine amino acids and urine organic acids confirmed OTCD diagnosis. The patient presented with severe hyperammonaemia (555 µmol/L, normal <60 µmol/L, acceptable <100 µmol/L) and liver failure and was hospitalised for emergency treatment. From day 1 of admission, she was treated for hyperammonaemia with enteral administration of lactulose (526 mg/kg/day), levocarnitine (88 mg/kg/day), L-arginine (250 mg/kg/day), carglumic acid (230 mg/kg/day), sodium phenylbutyrate (350 mg/kg/day) and sodium benzoate (250 mg/kg/day) and continuous haemodialysis (CHD; blood flow rate: 50 mL/min, dialysate flow rate: 6000 mL/hour) under intravenous sedation (midazolam 200 µg/kg/hour, fentanyl 1 µg/kg/hour) and intubated breathing control. The treatment reduced the blood ammonia concentration to an acceptable level (96 µmol/L) within 24 hours. She was weaned from CHD on day 8. Enteral citrulline (200 mg/kg/day) was added from day 11. The ammonia level remained within the acceptable range from 2–11 days. MRI performed on day 11 for assessment purposes showed symmetrical high-signal areas in the bilateral putamen (figure 1A) and no abnormality in the pons (figure 1B) on a T2-weighted image (repetition time (TR): 4940 ms, echo time (TE): 100 ms). A mitochondrial disease was suspected, but serum lactate and pyruvate levels were within normal ranges. The patient was extubated on day 12 and sedation ended; however, arousal was poor, and the ammonia level increased to >100 µmol/L. Sodium benzoate was switched to intravenous administration from day 13 (to day 20), and CHD (blood flow rate: 50 mL/min, dialysate flow rate: 2000 mL/hour) was resumed under intravenous sedation (midazolam 200 µg/kg/hour, fentanyl 1 µg/kg/hour) and intubated breathing management from day 14. The ammonia level decreased within the reference range on day 16; CHD, intubation and sedation were immediately terminated. The third hyperammonaemia observed on day 24 was improved by the doubling dose of enteral citrulline (400 mg/kg/day). Thereafter, ammonia levels remained normal, but mildly impaired consciousness persisted. Follow-up MRI on day 51 showed a characteristic trident-like high-signal area in the pons (figure 1D) besides the high-intensity areas in the bilateral putamen (figure 1C) on a T2-weighted image (TR: 4000 ms, TE: 98 ms). Based on these findings, the lesions in the bilateral putamen were considered to be EPM and the abnormality in the pons, CPM. The combined diagnosis was ODS. Regarding major osmolytes (Na+, Cl−, K+), no clinically problematic abrupt changes in serum levels were observed, Na+ (normal 135–143 mEq/L) and Cl− (normal 101–110 mEq/L) levels remained generally normal during treatment; however, hypokalaemia with a minimum value of 2.4 mEq/L (normal 3.6–4.9 mEq/L) was observed, coinciding with both sessions of CHD and intravenous sodium benzoate administration (online supplemental figure 1). Thereafter, the patient underwent liver transplantation and was discharged from the hospital; however, she still has mild consciousness disturbance and psychomotor retardation.
Supplemental material

{kind=link}
Serial T2-weighted magnetic resonance images of the brain on day 11 (A,B) and day 51 (C,D). (A) Symmetrical high-signal areas were observed in the bilateral putamen (arrows) on day 11. (B) No lesions were detected in the pons on day 11. (C) The high-signal areas in the bilateral putamen were still visible (arrows) on day 51. (D) A trident-like high-intensity area characteristic of central pontine myelinolysis appeared in the pons (arrowhead) on day 51.
Discussion
Osmotic pressure is the pressure at which a solvent moves from a solution of lower concentration to a solution of higher concentration when two chambers containing solutions of different concentrations are separated by a semipermeable membrane, that is, a membrane that allows only the passage of the solvent. The solute concentration in this context is referred to as osmolarity (in Osm/L). When a sudden change in osmolarity occurs in the environment around the cell, the cell is subjected to osmotic stress owing to the semipermeability of the cell membrane. Oligodendrocytes are particularly vulnerable to this stress, and rapid changes in osmolarity in the human body result in localised demyelination in the brain. This is the basic pathophysiology of ODS.4 5 10
However, it is difficult to merely explain our case of ODS secondary to hyperammonaemia treatment for OTCD and two previously reported similar cases with this factor alone, as shown below. Mattson et al and Cardenas and Bodensteiner did not attribute the aetiology to a direct change in osmolarity owing to ammonia correction, relying on the small molecular weight of ammonia.2 3 However, it is the amount of substance, not the molecular weight, that contributes to the osmolarity of solutions, as indicated by the following equation:
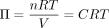
where Π is the osmotic pressure, n is the number of moles of the solute, R is the gas constant, T is the absolute temperature, V is the volume of the solution and C is the molar concentration of the solution (and in this context also specifically osmolarity).
We, therefore, calculated the osmolarity change rates directly owing to ammonia concentration change over the first 24 hours based on the substance amount in our case and the two similar cases mentioned above (although sufficient data were not available for the case of Mattson et al) and compared them with a consensus on the safe range of osmolarity change rates owing to serum Na+ concentration change (table 1).
Comparison of osmolarity change rates in each patient calculated by the changes in the serum ammonia concentration to a consensus on the safe range of the osmolarity change rates owing to serum Na+ concentration change
Na+ is best known as a solute whose rapid changes in serum concentration are responsible for the onset of ODS, and the consensus mentioned above is generally ±10 mOsm/L/day corresponding to ±10 mEq/L/day.5 12 Note that for the downward direction, –10 mOsm/L/day is quoted, which is the consensus for preventing the development of cerebral oedema since no consensus is established for preventing ODS development, although ODS due to the rapid descent of Na+ has also been reported.12 Since the resulting osmolarity change rates directly caused by ammonia correction (our case: –0.459, Cardenas and Bodensteiner3: –0.220, Mattson et al2: not less than –0.376 mOsm/L/day) were very small in absolute value compared with the consensus safe range for the rate of change in osmolarity by Na+, it is more natural to assume the existence of any other factor rather than to consider this as the sole cause of ODS.
We consider another factor to be the disruption of cell volume regulatory mechanisms, which may have been induced by hypokalaemia and hyperammonaemia. Cells have defence mechanisms against volume changes caused by osmolarity changes. Specifically, solutes are exchanged inside and outside the cell to provide homeostasis to the cell volume. The solutes that play this role in vivo are called osmolytes, the main osmolytes being Na+, Cl−, K+ and organic osmolytes such as glutamate and taurine.13 Although K+ has not received as much attention as Na+ in the context of ODS, hypokalaemia, which was also present in our case, was reportedly present in 66 of 74 cases of ODS secondary to the treatment of hyponatraemia,6 its presence was reported to worsen ODS prognosis7 and it alone could cause ODS.8 Its mechanism is currently unclear; however, we hypothesise that hypokalaemia might affect the smooth intracellular and extracellular exchange of K+, making cells vulnerable to osmolarity changes that are not normally problematic. Moreover, hyperammonaemia and liver failure (a well-known risk factor for ODS) were present in our patient. In this condition, glutamate decreases intracellularly and increases extracellularly in the brain to detoxify intracellular ammonia.14 Glutamate is an important organic osmolyte that contributes mainly to the reduction of intracellular osmolarity by being released from cells when cell volume needs to be reduced.13 In hyperammonaemia, the intracellular/extracellular concentration difference of glutamate can be reduced by the above mechanism, which may contribute to the disruption of cell volume regulatory mechanisms. Furthermore, most ammonia exists as NH4+ at physiological pH, which is known to compete with K+ in transport owing to its similar hydration radius and charge.15 This could affect volume regulation by K+.
Marked hyperammonaemia is frequently seen in patients with liver failure, a condition common in OTCD cases such as our case. Moreover, hypokalaemia can be caused by liver failure and following treatment with sodium benzoate (used in our case and Cardenas and Bodensteiner's3 case) or CHD (used in our case). Hence, patients with OTCD with hyperammonaemia and liver failure treated with sodium benzoate and CHD are at high risk of developing hypokalaemia and, consequently, ODS.
Early ODS diagnosis, primarily using MRI, is required to avoid serious consequences.4 9 The frequent use of MRI in recent years is regarded as a major factor in reducing ODS-associated mortality.4 The recent MRI finding that EPM precedes CPM, as in our case, suggests that MRI can further improve the prognosis of patients with ODS.9 11 Since the symptoms usually correspond to the lesion site,4 the presence of CPM rather than EPM is directly linked to poor prognosis, including locked-in syndrome, coma or death. If we could anticipate CPM based on EPM detection and correct the treatment course accordingly, the impact of this pathogenesis sequence on our daily practice would be considerable.
The differential diagnosis of high-signal areas in the bilateral putamen observed on T2-weighted images includes hypoxic encephalopathy, hypoglycaemia, methanol poisoning, mitochondrial diseases such as Leigh syndrome and EPM. However, based on the above discussion, we suggest that ODS should be suspected above all when the MRI of patients with OTCD treated for hyperammonaemia and presenting with hypokalaemia shows bilateral putaminal lesions or, more precisely, findings consistent with EPM.
As this was a single case report, cross-sectional retrospective observational studies are needed to accumulate further knowledge, for example, on the amount of time lag between EPM and CPM.
In conclusion, to our knowledge, this was the first report of EPM appearing before CPM in a case of ODS secondary to the treatment for hyperammonaemia due to OTCD. Our finding confirms the concept of ODS following the correction of hyperammonaemia in patients with OTCD, implying the importance of hyperammonaemia and hypokalaemia to understanding the pathophysiology of the condition and suggesting that MRI-based diagnosis may improve patient prognosis by anticipating the emergence of CPM when EPM is detected.
Data availability statement
Data sharing not applicable as no datasets generated and/or analysed for this study.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Jichi Medical University Central Clinical Research Ethics Committee approved this study (ID: 22-022). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors thank the patient, the patient’s family and the medical professionals who participated in managing this case.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors YS, SA and AK provided important insights into the patient’s condition. MM, RF, WN and HM contributed to the detailed interpretation of the clinical images. YT conceived and drafted the manuscript. All authors were involved in editing the manuscript and approved the final draft.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.