Article Text

Abstract
A rare extramedullary manifestation of haematological malignancy, myeloid sarcoma is most commonly seen in patients with acute myeloid leukaemia. We report on an adult patient who presented with an atypical phenotype of progressive cranial neuropathy without blood or bone marrow involvement, and in whom obtaining material for pathological diagnosis was made challenging by unusual findings of absent fluorodeoxyglucose-positron emission tomography avidity and involvement of sites not readily accessible to biopsy (orbital apex and cauda equina). The eventual diagnosis was obtained through biopsy of the uterine cervix before being verified on repeat lymph node and cerebrospinal fluid sampling prior to initiation of chemotherapy.
- clinical neurology
- histopathology
- neurooncology
- neuropathology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
We present a case of myeloid sarcoma presenting with multiple cranial neuropathies and polyradiculopathies, in which the diagnosis was ultimately made through biopsy of the uterine cervix. The case illustrates an unusual neurological presentation of a rare malignancy and highlights both the value of investigating incidental abnormalities and the need to reconsider working diagnoses when faced with the patient with progressive neurology.
Case report
A 63-year-old woman presented to hospital with a 1-week history of double vision and left eye pain. She had a history of diabetes mellitus, hypothyroidism and hypertension, and had been diagnosed with idiopathic aplastic anaemia 6 years previously, from which she had made a complete recovery.
Examination revealed a painful left-sided ophthalmoplegia, with pupil-involving third and left fourth cranial nerve palsies. Sixth cranial nerve function was intact. We found no other abnormalities on neurological examination.
CT and CT angiogram of the head were both unremarkable and MR scan of the brain and orbits was also reported as normal. We considered a diagnosis of a diabetic microvascular cranial neuropathy, but fourth cranial nerve involvement and pupillary involvement of the third cranial nerve seemed unusual for this diagnosis. A lumbar puncture was performed, demonstrating an acellular cerebrospinal fluid (CSF), and a mildly elevated CSF protein of 0.59 g/L (RR 0.15–0.45 g/L). Microbial cultures (including fungi) of the CSF were negative. Flow cytometry of peripheral blood was unremarkable, there was no detectable paraprotein and IgG4 levels were not raised. Extensive laboratory tests including angiotensin-converting enzyme, antinuclear antibodies, extractable nuclear antigen, anti-neutrophil cytoplasmic antibodies, HIV and syphilis serology were unremarkable. Erythrocyte sedimentation rate was not raised.
In light of these results, we suspected an inflammatory orbital apex syndrome and gave the patient prednisolone 50 mg daily, amitriptyline and pregabalin. There was rapid resolution of pain but no improvement in the ophthalmoplegia. Although aware that we had not established a definitive diagnosis, given the numerous negative investigations we adopted an expectant approach, with an early review scheduled for 2 weeks’ time.
On review 2 weeks later, the patient reported weakness and tingling of the right hand and numbness of the sole of the right foot. Examination revealed signs of a right C7 radiculopathy with moderate weakness of right elbow extension, mild weakness of right wrist, finger and thumb extension and a reduced right triceps jerk. In the lower limbs, the right ankle jerk was reduced and there was a sensory disturbance in the right S1 dermatome.
The combination of multiple cranial neuropathies and radiculopathies raised our suspicion for a meningeal disease process, and given her progressive neurology despite high-dose steroids we felt a neoplastic rather than inflammatory cause of her symptoms required further evaluation.
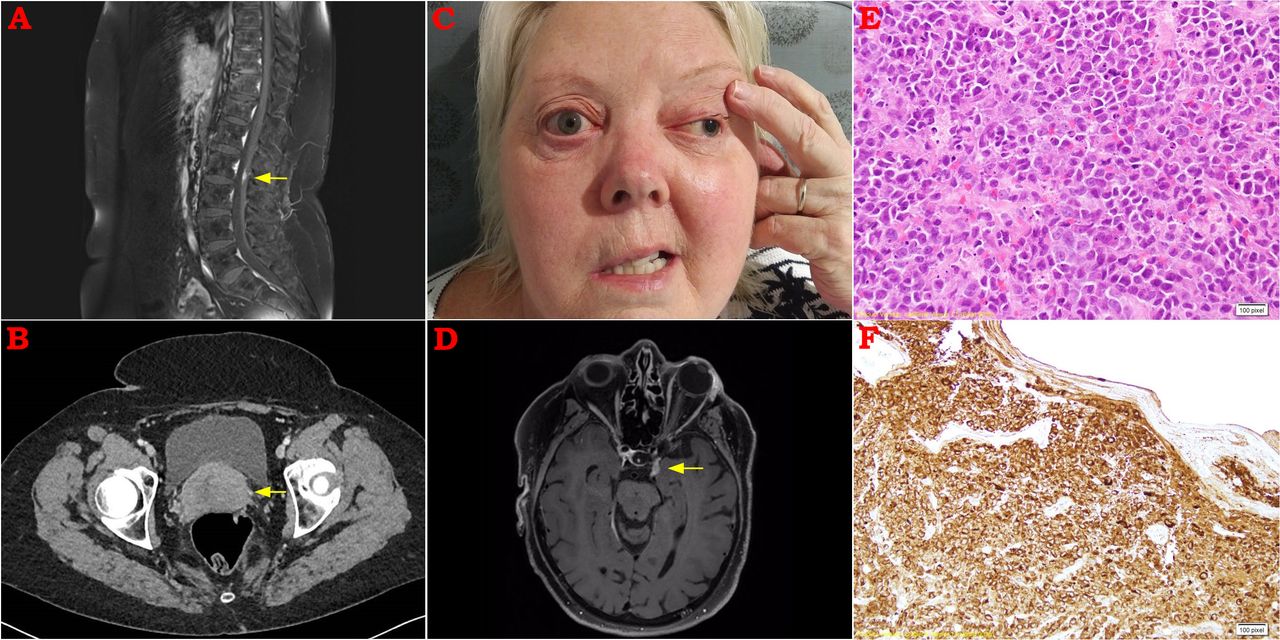
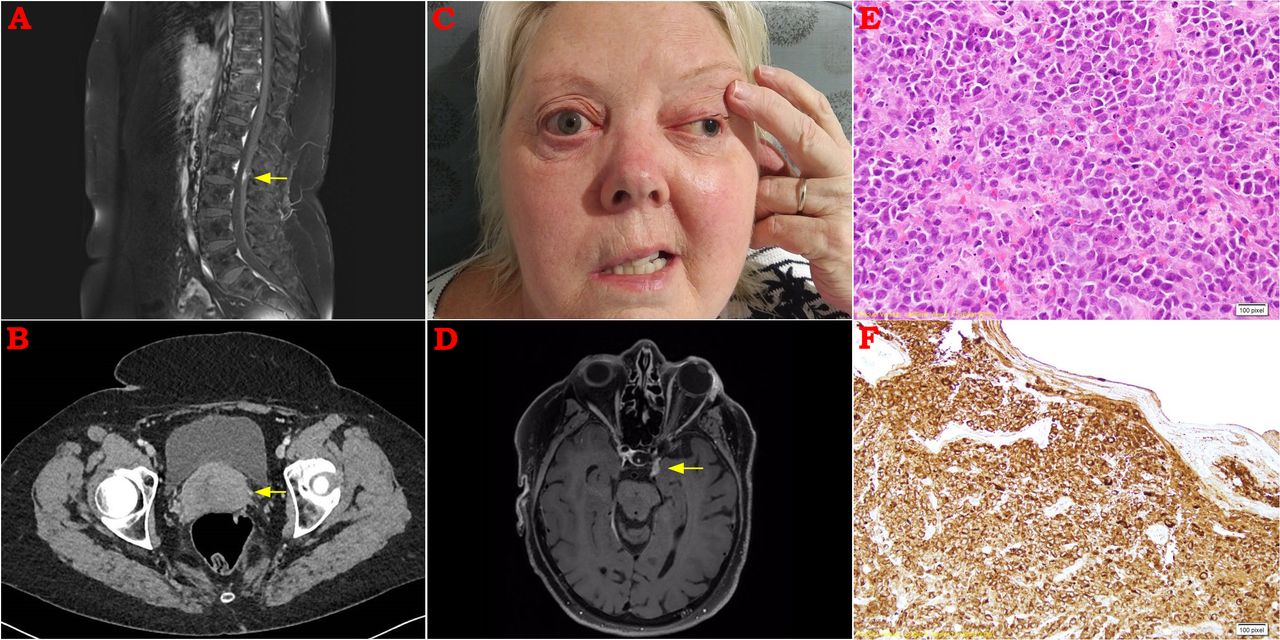
MR scan of the spine demonstrated a small region of nodular enhancement of the cauda equina (figure 1A). Full body fluorodeoxyglucose-positron emission tomography (FDG-PET) scan demonstrated no abnormal activity in any region. CT of the chest, abdomen and pelvis was normal apart from a bulky uterine cervix (figure 1B). At the time, we considered the cervical appearance to be an incidental finding and we arranged a gynaecological opinion. She continued high-dose prednisolone.
{kind=link}
(A) Postcontrast sagittal MRI spine demonstrating nodular enhancement among the nerve roots at the level of L2. (B) CT abdomen demonstrating well-defined bulky uterine cervix. (C) Clinical photograph demonstrating evidence of pupil-involving left cranial nerve (CN) III and right CN VII palsies. (D) Postcontrast axial MRI brain demonstrating cavernous sinus mass extending into left superior orbital fissure. (E) H&E stain of a biopsy from the uterine cervix demonstrating a cytologically malignant diffuse cellular infiltrate. (F) Immunohistochemistry was strongly positive for myeloperoxidase and CD68.
Three weeks later, she re-presented to hospital with difficulty swallowing, hoarse voice and left facial tingling. In addition to a left trigeminal sensory neuropathy, laryngoscopy revealed impaired right vocal cord function, consistent with a right 10th cranial nerve lesion. Several days later, she developed right facial weakness and signs of a right lower motor neuron facial nerve palsy (figure 1C). Although no difference in hearing was reported, audiometry revealed evidence of bilateral moderate to severe sensorineural hearing loss. Repeat CSF analysis demonstrated a protein of 1.26 g/L (RR 0.15–0.45 g/L) with 0.014×109 mononuclear cells (RR<0.005×109).
MRI was repeated, and demonstrated confluent enhancement of the cauda equina, leptomeningeal enhancement at the right temporal pole and abnormal soft tissue within the left cavernous sinus extending to the superior orbital fissure (figure 1D). Repeat CT scan of the abdomen showed new para-aortic lymphadenopathy, but a CT-guided core biopsy of the largest abdominal lymph node yielded minimal tissue and was non-diagnostic.
Although we had previously noted the abnormal appearance of the uterine cervix on CT abdomen, we had felt this to be an incidental finding, particularly as central nervous system involvement in cervical cancer is exceedingly rare.
Nonetheless, we pursued a biopsy of the bulky cervix which demonstrated diffuse proliferation of cytologically malignant cells, with immunohistochemistry staining strongly positive for myeloperoxidase and CD68, consistent with a myeloid sarcoma (figure 1E,F). Although bone marrow biopsy revealed no evidence of haematological malignancy, repeat lymph node and CSF sampling revealed immature myeloblasts confirming the diagnosis of an extramedullary haematological neoplasm.
The patient was transferred to a haematology unit, steroids were weaned and systemic chemotherapy was started. We repeated the FDG-PET scan (6 weeks after the first scan), and it showed widespread abnormal FDG uptake in the uterus and pelvic lymph nodes.
Unfortunately the patient developed chemotherapy-related complications, and following further deterioration with sepsis and seizures in the context of disease progression a palliative approach was adopted. Active treatment was withdrawn and the patient subsequently passed away.
Discussion
This case is of interest for a number of reasons. The first issue of interest is the pathological diagnosis. Myeloid sarcoma is a rare extramedullary tumour of immature myeloid cells that is most commonly seen in patients with acute myeloid leukaemia (AML).1 Nonetheless, myeloid sarcoma can also occur, as in the present case, in the absence of blood or bone marrow involvement. Diagnosis can be confirmed through tissue immunohistochemistry with staining for myeloperoxidase, CD43, CD68, lysozyme and CD117 positive in the majority of cases.2
If untreated, isolated myeloid sarcoma can transform to AML within 12 months and therefore early systemic chemotherapy is recommended even in the absence of leukaemic disease.3 To our knowledge, this is the first reported case of isolated myeloid sarcoma presenting with multifocal neurological manifestations and uterine involvement in an adult.
A second issue of interest is the fact that tissues with proven pathological involvement (lymph node and uterine cervix) were not initially FDG avid on the PET scan. This is despite previous case series reporting excellent utility of FDG-PET imaging for the detection of extramedullary disease in AML.4
There are several potential explanations for this false-negative FDG-PET result. First, the patient had been treated with high-dose corticosteroids for 5 weeks at the time of the first FDG-PET scan which may have suppressed metabolic activity.5 Second, the initial PET scan was performed relatively early in her presentation, when tumour burden was relatively mild. Six weeks later, as corticosteroids were being withdrawn and when tumour burden had advanced in both lymphatic and extralymphatic sites the FDG-PET scan was markedly abnormal.
Finally, this case illustrates the value of reinvestigation and reconsideration of a previous diagnosis when faced with progressive neurology. When our patient first presented with painful ophthalmoplegia involving the third and fourth cranial nerves with negative imaging of the brain and orbits, we concluded that an inflammatory orbital apex syndrome was the most likely diagnosis and treated accordingly. At that time, we also performed a range of investigations to exclude neurosarcoidosis, chronic meningitis, IgG4 disease and connective tissue disease.
When they subsequently developed multiple radiculopathies and cranial neuropathies, our suspicions of having missed an underlying meningeal malignancy were raised, however pathological confirmation remained elusive. We learnt an important lesson when the cervical biopsy revealed the diagnosis of myeloid sarcoma—that what may appear to be a ‘red herring’ or ‘incidentaloma’ on imaging is not always so.
Footnotes
Contributors All authors were involved in the patient’s case. AK drafted the manuscript. TK, PC and RG made critical revisions. All authors approved the final version to be published.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Obtained.
Provenance and peer review Not commissioned; internally peer reviewed.
Data availability statement Data sharing is not applicable as no data sets were generated and/or analysed for this study.