Article Text

Abstract
Patients with congestive myelopathy due to spinal dural arteriovenous fistula (SDAVF) typically present with progressive sensory and motor disturbance in association with sphincter dysfunction. Spinal MRI usually shows longitudinally extensive T2 signal change. Here, we report four patients with progressive myelopathy due to SDAVF who also presented with findings on cerebrospinal fluid (CSF) examination suggestive of an inflammatory aetiology. Such CSF findings in SDAVF are important to recognise, to avoid the erroneous diagnosis of an inflammatory myelitis and inappropriate treatment with immunosuppression. SDAVF can be difficult to detect and may require repeated investigation, with formal angiography as the gold standard.
- CSF
- MRI
- myelopathy
- neuroradiology
- neuropathology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Spinal dural arteriovenous fistula (SDAVF) can present with progressive gait, sensorimotor and sphincter disturbance due to a congestive myelopathy. Longitudinally extensive cord T2 signal change with T2 flow voids on MRI is diagnostic. In patients without typical clinical or imaging features, cerebrospinal fluid (CSF) examination may be performed to assess for inflammatory myelopathy, which can produce similar cord T2 signal change. While an ‘inflammatory’ CSF with pleocytosis and elevated protein in SDAVF is not well described in literature, we report four patients with such findings. This can result in initial misdiagnosis, unnecessary immunosuppression and delay in diagnostic investigations and appropriate treatment.1
Cases
Case 1
A man in his 60s presented with 1 week of progressive saddle anaesthesia, urinary retention and mild proximal leg weakness on a background of previous L3/L4 laminectomy. MRI demonstrated longitudinally extensive intramedullary T2 hyperintense signal change from T9 to the conus medullaris with mild spinal cord expansion. CSF revealed high protein of 1.25 g/L with pleocytosis (28 lymphocytes and six polymorphs/μL). The inflammatory CSF led to treatment with 3 days of intravenous methylprednisolone (1 g daily) with no symptomatic improvement.
Serial spinal MRI demonstrated progression of cord signal and expansion; postcontrast and time-resolved angiography with interleaved stochastic trajectories imaging revealed slightly prominent vessels posterior and to the left of the cord. Digital subtraction angiography (DSA) performed 2 months after symptom onset was unremarkable. Inflammatory CSF findings persisted (protein 0.93 g/L and lymphocytes 19/μL). Laboratory infective, vasculitic and paraneoplastic panels and aquaporin-4 and myelin oligodendrocyte glycoprotein antibodies were negative. Visual evoked potentials were normal. Electromyography demonstrated active denervation consistent with pathology proximal to the dorsal root ganglion. The patient subsequently received two doses of rituximab 700 mg for presumed inflammatory myelitis.
Despite immunosuppression, 6 months after symptom onset, the patient had persistent severe paraplegia and was wheelchair-bound. Repeat MRI demonstrated further proximal extension of signal change to T2 (figure 1A). CSF was persistently inflammatory (protein 1.61 g/L and 19 lymphocytes/μL). MRI of the brain and spinal dural biopsy results were normal. Repeat spinal DSA performed at 7 months after symptom onset under general anaesthesia revealed an SDAVF at left L3 (figure 1C). This was surgically resected via open laminectomy. His deficits improved following rehabilitation. At 12 months postresection, he had mild leg weakness, was mobilising with a frame and self-catheterising intermittently. Repeat spinal MRI 6 months postoperatively demonstrated resolution of T2 signal change (figure 1B).

(A) Case 1, sagittal T2 MRI demonstrating intramedullary hyperintensity to T2. (B) Case1, post-SDAVF resection sagittal T2 MRI demonstrating regression of hyperintensity to T6. (C) Case 1, spinal DSA demonstrating left L3 SDAVF. (D) Case 2, sagittal T2 MRI demonstrating intramedullary hyperintensity to T5. (E) Case 2, postembolisation sagittal T2 MRI demonstrating regression of intramedullary hyperintensity. (F) Case 2, spinal DSA demonstrating left L3 SDAVF. (G) Case 3, sagittal T2 MRI demonstrating intramedullary hyperintensity to T5. (H) Case 3, postembolisation sagittal T2 MRI demonstrating regression of intramedullary hyperintensity. (I) Case 3, spinal DSA demonstrating right L1 SDAVF. (J) Case 4, sagittal T2 MRI demonstrating intramedullary hyperintensity to T6. (K) Case 4, postresection sagittal T2 MRI demonstrating regression of intramedullary hyperintensity. (L) Case 4, spinal DSA with selective injection of left L1 segmental spinal artery showing a left L1 SDAVF in the typical location of the nerve root dura under the left L1 pedicle. The fistula is supplied by dural branch and drains to congested ascending medullary veins. DSA, digital subtraction angiography; SDAVF, spinal dural arteriovenous fistula.
Case 2
A man in his 60s presented with a 9-week history of progressive leg weakness, distal dysaesthesia and urinary retention. Lower limb examination revealed severe diffuse asymmetric paraparesis, absent reflexes and patchy sensory loss. Spinal MRI at admission showed longitudinally extensive intramedullary high T2 signal from T5 to the conus with mild contrast enhancement of the cauda equina and swelling of nerve roots, but without abnormal intradural flow voids (figure 1D).
CSF performed on three occasions over 8 weeks previously and at admission showed persistent lymphocytic pleocytosis (range 11–93/μL), elevated protein (range 1.46–1.9 g/L) with normal glucose and negative oligoclonal bands. Electromyography revealed fibrillations in the lumbar myotomes.
Spinal DSA, performed 10 weeks after admission, failed to identify a spinal vascular malformation. Left L5 nerve root biopsy showed patchy axonal and myelin degeneration without evidence of lymphoma.
He was later diagnosed with Cowden’s syndrome, an autosomal dominant multisystem hamartomatous disease due to PTEN mutation associated with peripheral and central vascular malformation. This led to a second spinal DSA performed under general anaesthesia 20 weeks from symptom onset, demonstrating left L3 SDAVF (figure 1F), which was embolised with histoacryl. At 4 months’ follow-up he had mild asymmetric distal leg weakness with persistent sphincter dysfunction. Spinal MRI demonstrated a significant reduction in cord oedema (figure 1E). This case was reported in detail elsewhere.2
Case 3
A middle-aged patient was admitted with a 3-month history of progressive paraparesis, patchy dysaesthesia and sphincter dysfunction. Lower limb examination demonstrated moderate asymmetric weakness. Spinal MRI showed a longitudinally extensive T2 signal change from T5 to conus with several possible flow voids (figure 1G). CSF on admission was pleocytic (140 lymphocytes/μL) with elevated protein (1.95 g/L). Electromyography revealed fibrillations in the lumbar myotomes. Spinal DSA under local anaesthesia revealed a right L1 SDAVF (figure 1I), which was embolised with histoacryl.
At 3 months’ follow-up, sphincter dysfunction had resolved, and knee jerks returned. Mild weakness of the right ankle dorsiflexion and toe extension persisted. Follow-up spinal MRI (figure 1H) showed a nearly complete resolution of cord signal change.
Case 4
A man in his 60s presented with 12 months of slowly progressive asymmetric leg weakness followed by reduced leg and perianal sensation. There was then a rapid deterioration with paraparesis, paraesthesia and bilateral thigh pain, progressing to urinary retention and faecal incontinence. MRI spine with contrast demonstrated a faintly enhancing conus lesion with associated mainly posterior cord signal change extending to T6, suspicious for low-grade astrocytoma or inflammatory myelopathy (figure 1J).
CSF showed 8 lymphocytes/μL, elevated protein of 1.99 g/L with no unmatched oligoclonal bands. Cytology and flow cytometry revealed no atypical cells. Due to concern about malignancy, he underwent T12 laminectomy and biopsy of the cord lesion. Histopathology showed no definitive tumour, although an infiltrating edge of a low-grade glioma could not be excluded by the histopathologists.
Repeat MRI demonstrated the mildly enhancing terminal cord lesion with oedema and signal change extending up to the thoracic cord. Given the inflammatory CSF, oral dexamethasone 4 mg four times per day was commenced with mild radiological improvement in degree of swelling, but there was ongoing clinical deterioration.
The patient was transferred to our hospital for further management. At this point, 3 months after acute deterioration, power was Medical Research Council grade 2/5 in hip flexion, 4+/5 in hip extension, 2/5 in knee flexion, 4+/5 in knee extension and 4−/5 in dorsiflexion. Reflexes were absent and plantars were mute. Proprioception and pinprick sensation were felt at the ankles and vibration was felt at the knees. He was wheelchair-bound and needed mechanical assistance to stand.
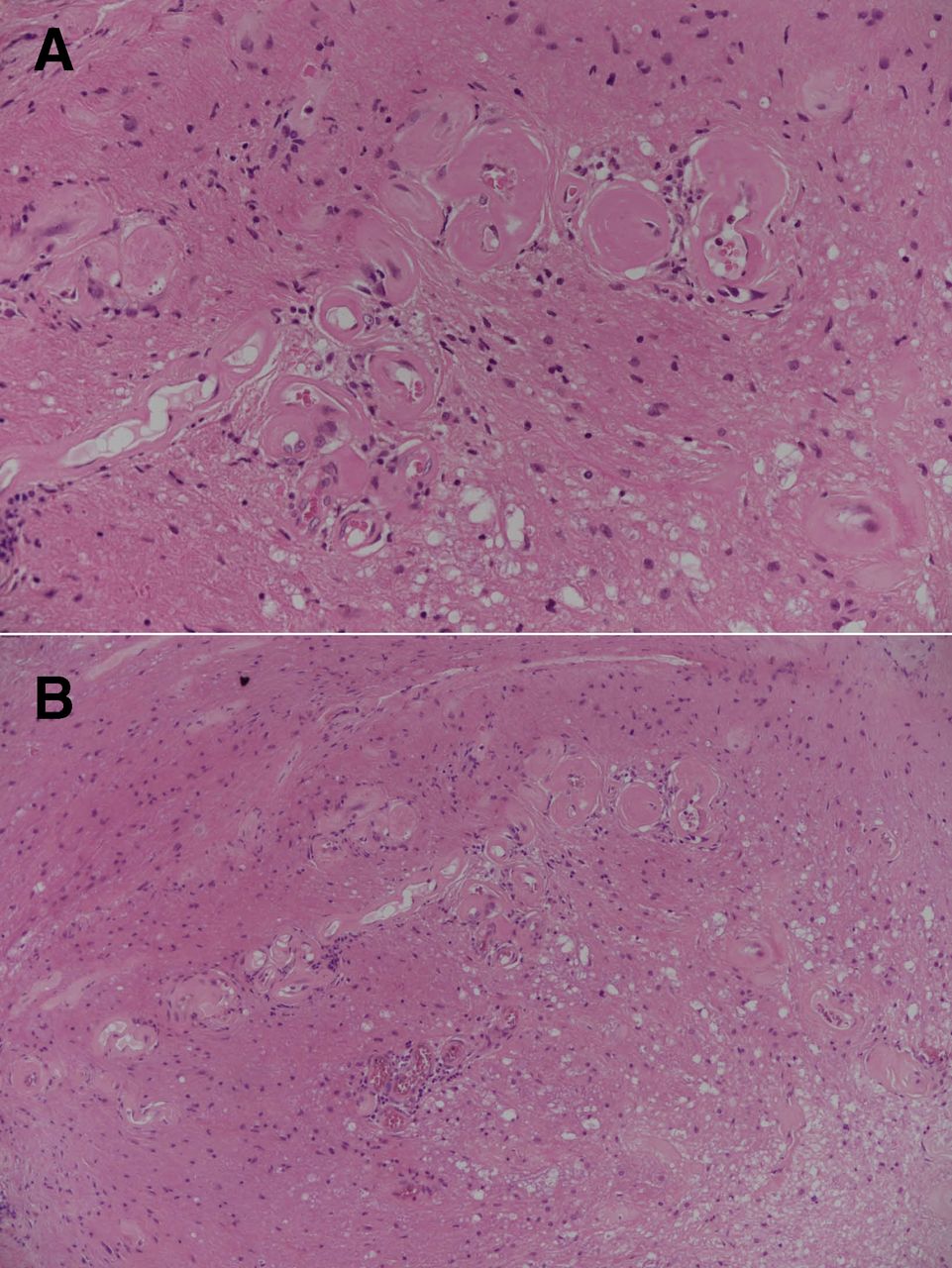
Repeat CSF was again pleocytic (120 polymorphs and 25 monocytes/μL) with high protein (1.83 g/L). Cytology was suggestive of a reactive, acute inflammatory process with no atypical cells. Histopathology from the spinal cord biopsy was re-reviewed. There was no evidence of neoplasia, but presence of abnormal parenchymal vascularity with increased, and hyalinised small vessels were in keeping with SDAVF and secondary myelopathic changes (Foix-Alajouanine syndrome) (figure 2). Subsequent DSA under general anaesthesia confirmed a left L1 SDAVF (figure 1L). He underwent surgical disconnection of the fistula with clinical improvement. Twelve months postoperatively, he could ambulate with a walking aid and was managing intermittent self-catheterisation. There was also radiological improvement in the degree of cord signal change (figure 1K).
{kind=link}
{kind=link}
Histology from spinal cord biopsy at high (A) and medium (B) power shows abnormal vascularity in the spinal cord parenchyma with an increased number of hyalinised small vessels in keeping with a spinal dural arteriovenous fistula. In addition, there are secondary myelopathic changes. These findings are compatible with Foix-Alajouanine syndrome.
Discussion
A longitudinal extensive myelopathy with CSF pleocytosis usually suggests inflammatory demyelinating aetiologies but can rarely be due to SDAVF. While raised CSF protein is a common finding seen in up to three-quarters of patients,3 presence of pleocytosis is under-recognised and may result in initial misdiagnosis and treatment with steroids, which can worsen cord oedema.
Raised CSF protein and pleocytosis in SDAVF may be related to venous hypertension from an arterialised venous drainage system resulting in raised intravenule pressure,4 5 with transudative flow of protein and cells into the extravenule space and CSF. Kilic et al reported presence of mature lymphocytes in a patient with SDAVF also treated initially with steroids, but detailed CSF analysis was not described.1 To our knowledge, this is the largest case series reporting inflammatory CSF in SDAVF.
These four patients with SDAVF were managed initially with steroids due to inflammatory CSF, along with non-diagnostic imaging or initial DSA. These cases suggest that such CSF findings should not discourage clinicians from pursuing a diagnosis of SDAVF in the appropriate clinical context.
While spinal flow voids on MRI may assist in diagnosis, these changes are often subtle and are not universally present.6 Spinal DSA remains the gold standard in diagnosis of SDAVF, but the procedure can be difficult due to anatomical challenges and patient tolerance.7 In the case of patients 1 and 2, a second procedure under general anaesthesia was required. Repeat DSA is important in patients with clinicoradiological features consistent with SDAVF.
Our report suggests that inflammatory CSF may be associated with SDAVF and should not deter clinicians from investigating for this treatable condition.
Footnotes
Twitter @teddyyhwu, @AndrewHEv
Contributors VV and TW had the idea for the article. VV and VL performed the literature search and wrote the article. RD, RHR, BJMcG, DHK, HM, AQ, MT, FR, WT, AE, SB and PM identified and/or managed the cases, and edited and revised the manuscript for intellectual content. All authors approved the final version. VV is the guarantor.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Obtained.
Provenance and peer review Not commissioned; internally peer reviewed.
Data availability statement No data are available.